

 新火種
2024-11-27
新火種
2024-11-27 


高精度預測蛋白構象變化,中國科大、上科大通用深度學習模型
編輯 | KX
預測蛋白質構象變化是計算生物學和人工智能領域的一大挑戰。主流的 AlphaFold 等算法可以高通量預測蛋白質的靜態結構,但對蛋白質構象變化預測卻束手無策。
為了解決這個問題,中國科學技術大學和上海科技大學的研究人員,提出了一種新穎的深度學習策略,即利用高通量生物物理采樣來規避與蛋白質構象轉變相關的數據匱乏。
研究人員將分子動力學模擬與增強采樣方法相結合,創建了一個大規模數據庫。研究模擬了 2635 種已知兩種穩定狀態的蛋白質的構象變化,并收集了每條轉變途徑的結構信息。利用這個數據庫,開發了一個能夠預測給定蛋白質轉變途徑的通用深度學習模型。
該模型對不同序列長度(從 44 到 704 個氨基酸)的蛋白質表現出普遍的穩健性,并適應不同類型的構象變化。在幾個系統中,預測和實驗數據結果高度一致,并成功地應用該模型來識別一個重要生物系統——人類 β-心臟肌球蛋白中的一種新的變構調控。
這些結果證明了該模型在揭示蛋白質構象變化本質方面的有效性。
相關研究以「Exploring Protein Conformational Changes Using a Large-Scale Biophysical Sampling Augmented Deep Learning Strategy」為題,發表在《Advanced Science》上。

AlphaFold 和 RoseTTAFold 等致力于預測蛋白質的靜態結構,但并未解決大多數蛋白質在發揮其生物學作用時所經歷的動態構象變化。
這些轉變對于理解從酶活性到信號轉導的廣泛生物過程至關重要。然而,缺乏中間狀態的結構數據阻礙了對這些轉變的預測。此外,現有模型的過渡態自由能壘較高,使得準確預測更具挑戰性。
現有的描述蛋白質構象轉變的模型包括基于彈性網絡的正常模式分析,以及將彈性網絡與分子動力學模擬相結合的混合模型。這些方法適用于相當簡單的構象運動,但無法解釋在較大的蛋白質中發現的復雜和巨大的變化。
最近,深度學習方法將蛋白質結構映射到低維潛在空間。然而,這些模型依賴于兩種狀態之間的線性路徑,這不適用于復雜的非線性轉換,例如倍數切換。更重要的是,高數據需求和低數據效率,以及阻礙實時可擴展應用的計算成本,使得這些方法本身無法令人滿意。
模擬 2635 種蛋白質的構象變化受深度學習在預測靜態蛋白質結構方面的成功啟發,研究人員現在正在積極探索其他旨在預測蛋白質構象變化的深度學習算法。其主要挑戰在于表征不同構象轉變的訓練數據有限。
在此,中科大團隊采取了更直接的方法來彌補數據短缺的問題。通過結合分子動力學模擬和增強采樣方法,廣泛模擬了 2635 種蛋白質的構象變化,其中包含兩種實驗確定的狀態,即單狀態 (SS) 和多狀態 (MS) ,并直接獲得了組成轉換途徑的結構。
SS 和 MS 蛋白質數據集的創建涉及對蛋白質數據庫 (PDB) 中的結構進行全面搜索和比較。
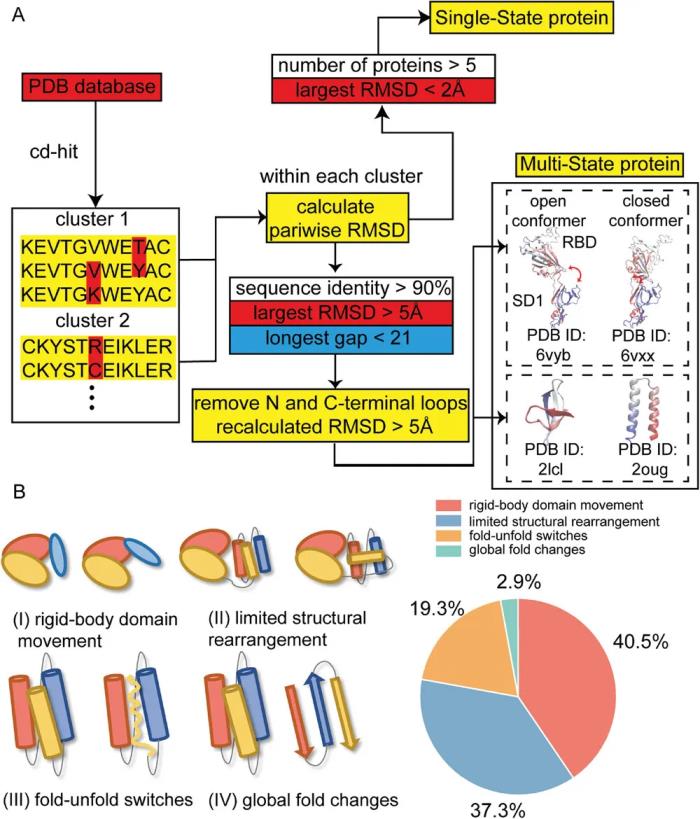
SS 數據集共包含 3454 個條目,包括在實驗研究中觀察到的具有高度收斂構象的蛋白質。相比之下,MS 數據集包含 2635 種蛋白質(每種蛋白質在 PDB 中都有兩種結構),這些蛋白質表現出顯著的構象變化,定義為均方根偏差 (RMSD) >5 ?。
該數據集可進一步分為四個子類:剛體結構域移動(rigid-body domain movement)、有限的結構重排(limited structural rearrangement)、折疊-展開切換(fold-unfold switches)、全局折疊變化(global fold changes )。
高精度預測蛋白轉變途徑的通用 AI 模型該數據集使用一種稱為 PATHpre 的整體深度學習模型,可以高精度地預測導致構象轉變的結構途徑。
PATHpre 中 HESpre 模塊的核心創新在于預測轉變途徑中高能狀態的性能。所提出的模型對不同構象的蛋白質具有極大的通用性。這意味著它對復雜系統中的動力學行為建模做出了巨大貢獻,在方法級別應用了可擴展性和數據效率。
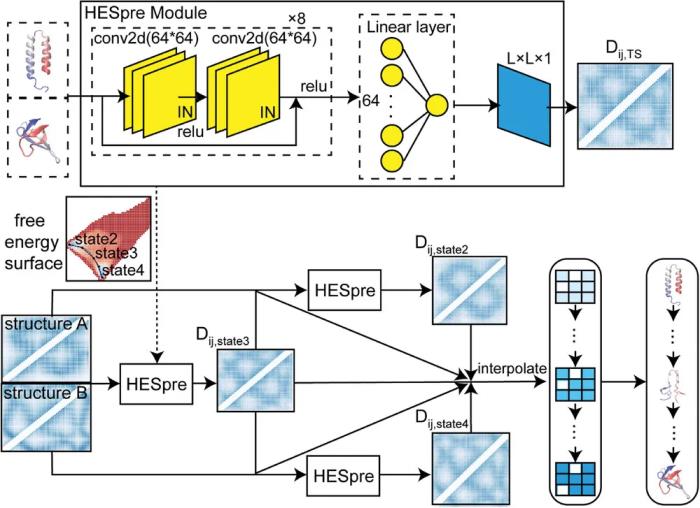
在 PATHpre 方法中,通過卷積神經網絡預測應用兩種構象狀態系統中的距離矩陣,以獲得這些構象狀態之間的高能狀態。核心模塊名為 HESpre,目的是預測高能狀態下的結構信息。然后迭代使用該模塊來預測整個途徑。
研究對各種蛋白質進行了交叉驗證,該模型在所有步驟中都實現了強大的 Pearson 相關性和低平均絕對誤差;因此,它在結構類別中非常通用。
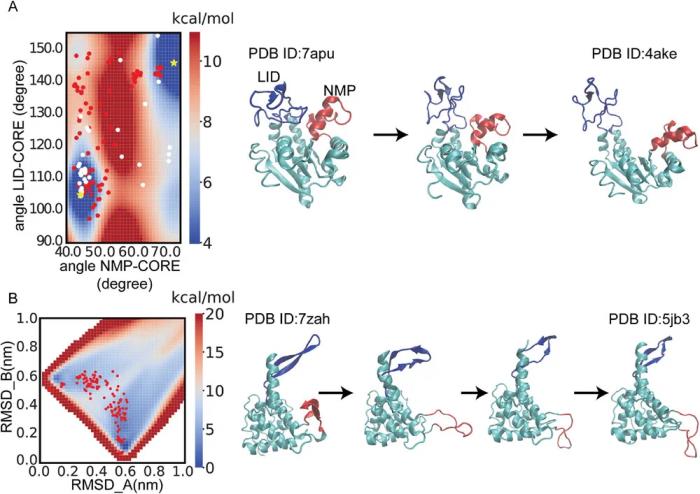
PATHpre 通過與一系列蛋白質上存在的實驗和模擬數據顯示出強相關性,在蛋白質轉變途徑非常高的預測中很準確。評估還表明,PATHpre 可以穩健地捕獲從簡單到復雜的構象變化,并且它與不同長度的序列以及結構復雜性保持一致。
重要的是,它通過匹配實驗自由能景觀,準確預測了單個蛋白質(如腺苷酸激酶和 30S 核糖體蛋白 S7)的轉變途徑,并且在具有挑戰性的條件下,比傳統的混合方法表現更好。PATHpre 的預測與已知結構一致,其對折疊轉變蛋白質中精細中間狀態的映射證實了其廣泛的適用性和可靠性,可以捕獲廣泛的蛋白質構象轉變。
蛋白質構象變化新見解與其它幾種同類蛋白質構象變化深度學習模型相比。PATHpre 模型具有以下特點:
首先,PATHpre 使用來自模擬的轉變數據進行訓練,消除了轉變路徑在潛在空間中呈線性的假設。
其次,模型是適用于所有蛋白質的通用模型,無需針對每種蛋白質進行自定義再訓練。
第三,模型專注于結構明確的蛋白質的大構象變化(RMSD > 5?),而不是內在無序的蛋白質或局部變化相對較小的蛋白質,這在藥物設計中很重要。
該研究標志著 AI 驅動的蛋白質建模取得了重大進展,為預測蛋白質構象變化提供了一種數據高效且可擴展的方法。該模型將進一步提高科學家對蛋白質構象變化的理解。
數據庫和源代碼地址:https://github.com/qwang897/PATHpre
參考內容:https://www.marktechpost.com/2024/11/03/a-study-on-protein-conformational-changes-using-a-large-scale-biophysical-sampling-augmented-deep-learning-strategy/
相關推薦



- 免責聲明
- 本文所包含的觀點僅代表作者個人看法,不代表新火種的觀點。在新火種上獲取的所有信息均不應被視為投資建議。新火種對本文可能提及或鏈接的任何項目不表示認可。 交易和投資涉及高風險,讀者在采取與本文內容相關的任何行動之前,請務必進行充分的盡職調查。最終的決策應該基于您自己的獨立判斷。新火種不對因依賴本文觀點而產生的任何金錢損失負任何責任。
