

 新火種
2024-11-17
新火種
2024-11-17 


百萬級原子模擬,從頭算精度,北京科學智能研究院提出AI+大尺度電子結構模擬新方法

編輯 | KX
在計算材料科學領域,準確高效地模擬材料的電子結構一直是一個非常關鍵而又極具挑戰性的問題。基于密度泛函理論的第一性原理計算方法的高計算需求依然是大尺寸長時間材料模擬所面臨的難題。
北京科學智能研究院 (AI for Science Institute, Beijing) 提出了一種基于深度學習的高效緊束縛方法,稱為 DeePTB,從而高效地表示具有從頭算精度的材料電子結構,極大地簡化了計算復雜度,并實現百萬級大尺寸結構的電子、光電響應性質的計算模擬。
當與分子動力學相結合時,DeePTB 可以同時促進原子和電子行為的有效和準確的有限溫度模擬。DeePTB 的可用性彌合了電子模擬中準確性和可擴展性之間的差距,通過實現大規模電子結構計算,將推動材料科學和相關領域的發展。
相關研究以「Deep learning tight-binding approach for large-scale electronic simulations at finite temperatures with ab initio accuracy」為題,于 8 月 8 日發表在《Nature Communications》上。

雖然基于 DFT 的第一性原理方法提供了準確且通用的模擬材料電子性質的方法,但是隨著系統中的原子數量增加,第一性原理的計算量急劇增加,而在真實材料或者器件體系中往往包含成百萬千萬量級的原子數,難以很直接使用第一性原理軟件完成計算模擬。
一些復雜材料場景遠遠超過了 DFT 方法的模擬尺寸,一方面是因為 DFT 的自洽迭代過程復雜,另一方面,DFT 需要足夠大的基組來保證精度,導致產生的哈密頓量的尺寸較大,難以進行后續的性質計算。
因此使用更小和更稀疏的矩陣來描述電子哈密頓量的緊束縛(Tight-Binding, TB)方法提供了一種更為實用的替代方案。然而,傳統的 TB 方法也存在精度與效率的矛盾。例如基于 Wannier 函數的 TB 方法雖然具有較高的精度,但是其構造過程需要 DFT 自洽迭代計算,仍然限制了其在大尺寸體系下的應用。
因此發展通用的 TB 哈密頓量模型方法軟件框架 DeePTB 實現精度效率以及遷移率的統一,是具有重要意義的課題。
DeePTB 方法框架及特點DeePTB 方法的整體框架圖下圖所示,對于給定構型中指定近鄰范圍內的成鍵原子對, 首先訓練其基于物理約束下的經驗公式系數,然后在此基礎上提取成鍵原子對的局域化學環境,通過 embedding 網絡構造 symmetry-preserving 描述子,并基于 fitting 網絡映射為局域環境依賴的 TB 參數,突破傳統模型的雙中心近似,并基于 PyTorch 機器學習框架,利用 DFT 電子本征值構造損失函數,系統地進行高效的參數自動擬合。
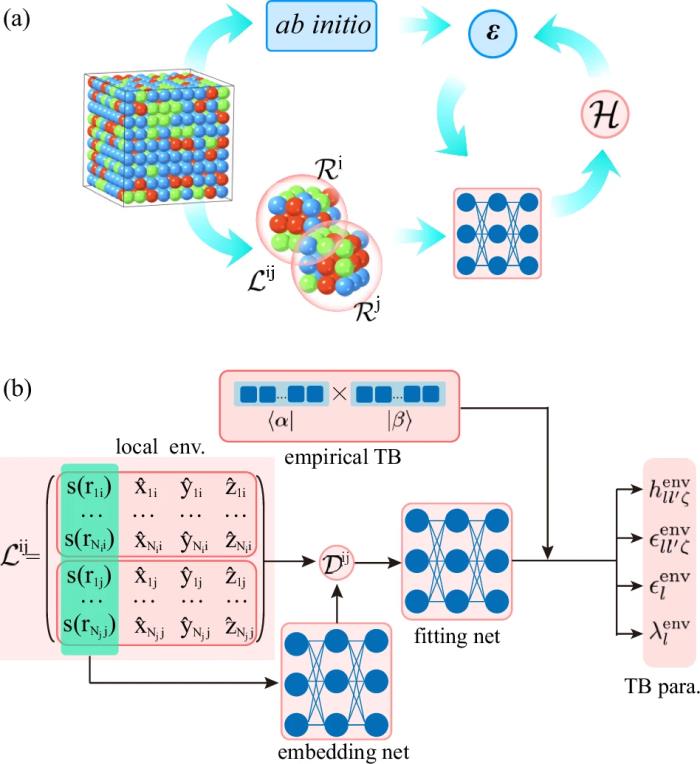
DeePTB 方法具有如下特點:
實現精度與效率的統一,通過神經網絡修正實現以經驗緊束縛模型計算效率并保持第一性原理的計算精度。采用 eigenvalues 作為訓練標簽,用戶可以靈活地選擇任何 DFT 軟件生產訓練標簽,可以是平面波基組,也可以是LCAO 基組,也可以是任意泛函(LDA、GGA、even Hybrid functionals)。同時也可以輕松實現并處理自旋軌道耦合相互作用。使用更小的基組,相對完整的 LCAO DFT 哈密頓量,TB 使用的更小的基組,甚至做到只擬合費米面附近的能帶。采用正交基組 TB 形式,無需額外處理交疊矩陣,因此可以接入大規模 TB 算法,例如 tight-binding propagation method (TBPM) ,輕松實現百萬千萬量級原子的第一性原理精度的電子性質計算。真正實現器件級尺寸的量子力學模擬。基于 Slater-Koster 框架,支持用戶自定義經驗 TB 擬合公式,并可以系統地增加神經網絡修正,提高精度。為目前文獻上存在各種經驗擬合公式以及參數提供一個統一的實現和提高精度的訓練平臺。支持與分子動力學結合,實現有限溫度下原子的動力學過程中以及結構系綜采樣中的電子結構和性質的模擬。預測結構擾動構型的 TB 哈密頓量以及電子結構研究人員以在電子器件中被廣泛使用的 IV 族元素(C、Si、Ge、Sn)和 III-V 族化合物(如 GaAs 等)組成的半導體材料作為測試對象。
首先,進行分子動力學(MD)模擬在有限溫下的結構構型采樣,并基于不同 MD 軌跡的構型,使用 DFT 軟件計算其對應的電子本征值作為 DeePTB 的訓練和測試數據。模型測試全部體系的決定系數 (R^2≈0.9999 ) ,本征值偏差只有十幾至幾十個 meV 左右。其中 III-V 族化合物的測試集同時包含了立方和六方兩種不同的相下的構型。
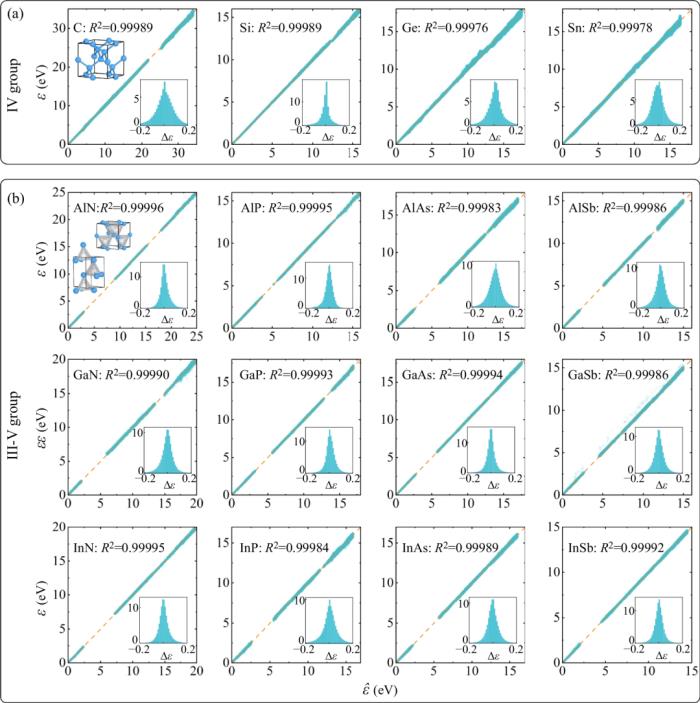
此外,DeePTB 模型還展現出了以下出色的泛化能力:
推廣到更大尺寸的超胞結構,顯示出極佳的尺度可擴展性。處理應變效應,準確預測應變調控下的能帶結構及帶隙大小。兼容不同的 DFT 基組、泛函和自旋軌道耦合效應,表現出強大的靈活性和通用性。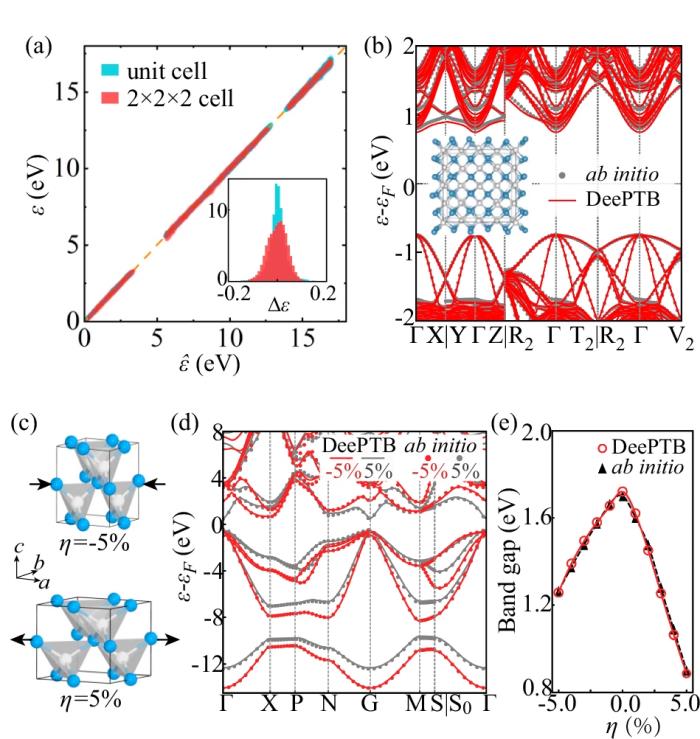
研究人員選擇 III-V 族化合物 GaP 作為大尺寸建模的應用案例,構造了 50 × 50 × 50 的超胞結構。
首先基于 DP 深度勢能進行 DeePMD 分子動力學模擬有限溫的結構采樣,然后基于得到的采樣構型,利用 DeePTB 進行緊束縛模型哈密頓量的構建,并基于預測的 TB 模型使用 TBPLaS 軟件實現的 TB propagation method (TBPM) 方法進行無需對角化的快速的電子性質計算,得到包括有限溫下的態密度(DOS)、光電導率、介電函數以及復折射率等電子性質及光電相應,如下圖所示。
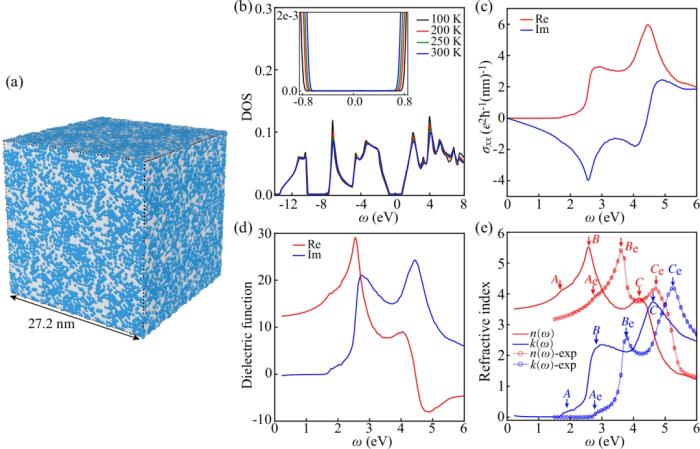
計算結果表明, DeePTB 的計算結果與文獻結果符合良好,峰值位置的輕微差異主要是因為用于訓練的 DeePTB 模型的交換關聯泛函(GGA)傾向于低估半導體材料的電子帶隙的緣故。這些結果表明了 DeePTB 高精度建模以及進行器件級尺度電子結構及性質的模擬計算的能力。
關于 DeePTB 框架的潛力對于不同的交換關聯 (XC) 函數,能帶結構的色散特征大致相同。因此,原則上,可以首先在計算效率高的 XC 函數(如 LDA 或 GGA)上訓練模型,然后將其轉移到更昂貴、更準確的函數(如 SCAN 或 HSE)。這使得能夠高度準確地描述實驗可觀測量,以用于接近現實的材料模擬等情況下所需的大規模模擬。
此外,對于大規模樣本,模擬應變對電子特性的影響是一項計算繁瑣的任務。DeePTB 可以通過在較小的樣本上訓練模型并將其轉移到更大的系統來有效地加速這些模擬。這為電子結構應變工程的理論研究帶來了優勢。
MD 可以提供離子自由度的模擬,這類似于晶體結構的溫度探針,其中離子振動是基本現實。在需要大規模和長時間模擬的情況下,DeePTB 可用于模擬溫度和結構相關的電子特性。DeePTB 使得考慮其他實際情況(如缺陷或雜質及其對電子結構的影響)成為可能和可行。
DeePTB 探索的另一個方向是模擬磁系統的特性。鑒于 DeePTB 的這些多樣化潛在應用,它可以在電子模擬領域產生深遠的影響。
參考內容:https://mp.weixin.qq.com/s/StetT81-UD6AGGgv-60GPA
相關推薦


- 免責聲明
- 本文所包含的觀點僅代表作者個人看法,不代表新火種的觀點。在新火種上獲取的所有信息均不應被視為投資建議。新火種對本文可能提及或鏈接的任何項目不表示認可。 交易和投資涉及高風險,讀者在采取與本文內容相關的任何行動之前,請務必進行充分的盡職調查。最終的決策應該基于您自己的獨立判斷。新火種不對因依賴本文觀點而產生的任何金錢損失負任何責任。
