

 新火種
2024-04-11
新火種
2024-04-11 


專訪騰訊AILab姚建華、楊帆:騰訊AILab為何瞄準單細胞蛋白質組學?
在生物醫學研究的前沿領域,“單細胞蛋白質組學”是怎樣的存在?
用一個比喻來說,它就像一把鑰匙,能夠開啟細胞內部世界的大門,讓我們得以窺見細胞如何通過蛋白質的相互作用來執行生命活動。
這一研究領域的突破,不僅能夠推動科學界對生命過程的理解,也為精準醫療的實現奠定了基礎。
近期,騰訊的 AI Lab,無疑成為了這一前沿研究領域率先“揭開英雄榜 ”的那個研究機構。
3月20日,騰訊 AI Lab 的 3 篇蛋白質組論文正式入選國際頂級學術期刊。論文分別在數據庫、AI 建模、AI 輔助臨床三個角度提出了全新的研究方案,為人類從根本上闡釋生命提供了重要技術參考。
《SPDB: a comprehensive resource and knowledgebase for proteomic data at the single-cell resolution》,被生物信息學領域數據庫方面的的權威期刊 Nucleic Acids Research收錄。
《 scPROTEIN: a versatile deep graph contrastive learning framework for single-cell proteomics embedding》,被Nature旗下的方法學期刊Nature Methods收錄。
《Deep domain adversarial neural network for the deconvolution of cell type mixtures in tissue proteome profiling》,被Nature旗下機器學習專業期刊 Nature Machine Intelligence 所收錄。
借此契機,近期對話騰訊 AI Lab 科學家姚建華和研究員楊帆,他們是三篇論文的共同作者。在訪談中,他們深入闡述了這些論文背后的技術突破、應用價值和未來的研究規劃。
他們解釋道,這三篇論文的創新之處在于,它們首次為單細胞蛋白質組提供了全面的數據知識庫和系統的AI分析方法。
論文一中建立的 SPDB 數據庫,通過標準化處理不同來源的單細胞蛋白質組學數據,使得數據易于比較和分析,是目前全球數據量最大、覆蓋技術和數據集最為廣泛的單細胞蛋白質數據庫。
論文二中的 scPROTEIN 框架,針對單細胞蛋白組數據的特殊性提出了解決方案,能夠處理數據中的不確定性、缺失值、批次效應和噪聲問題。為基于單細胞蛋白質組的腫瘤發生發展機制研究、藥物靶點發現和腫瘤早篩和微環境研究提供重要的AI輔助作用。
第三篇論文中提出的 scpDeconv 方法,是一種全新的反卷積方法,能夠從“組織蛋白質組”數據中挖掘出特定細胞類型比例,為腫瘤輔診和預后分析提供了新的視角,是三篇論文中與臨床應用最為貼近的一項成果。
姚建華,作為騰訊 AI Lab 的 AI 醫療首席科學家,補充道:
“AlphaFold 在蛋白質結構領域取得了令人矚目的成就,它主要關注單個蛋白質的結構和功能,或幾個蛋白質之間的相互作用。
而我們的研究則聚焦于細胞內所有蛋白質的表達模式,這些信息反映了整個細胞的狀態和微環境,使我們的工作更加貼近臨床應用和疾病機制的探索。”
值得一提的是,當我們在討論論文成果的同時,一個更深遠的議題逐漸浮現:成立于2016年的騰訊 AI Lab,是否有能力在接下來的五年中,引領生命科學領域的未來發展?
這個問題不僅考驗著實驗室的科研實力,也反映出科技公司在生物醫學領域的影響力和責任。如今的騰訊 AI Lab,走的每一步都比以往更受關注。
以下為對話(經編輯):
數據、建模、應用,「三管齊下」
楊帆:單細胞測序技術已經取得了飛速發展,盡管單細胞轉錄組相關的測序技術和計算方法已經相當成熟,但轉錄水平與蛋白質水平的相關性通常低于 50% 。在單細胞層面,這種相關性更低。
因此,只有通過研究蛋白質組,我們才能深入理解生命活動和疾病的本質。
單細胞蛋白質組測序技術也在不斷進步,技術革新層出不窮,并受到了國際頂級期刊如 Nature Methods 的關注和報道。特別是以 SCOPE-MS(Single-Cell Proteomics by Mass Spectrometry)、nanoPOTS (nanodroplet processing in one pot for trace samples) 為代表的基于質譜的蛋白質測序技術,能夠檢測到單細胞中數千種蛋白質的存在。這比以往基于抗體的單細胞蛋白質組測序技術有了顯著的提升。
然而,這些數據的復雜性,使得專門針對單細胞蛋白質組數據的AI計算方法相對缺乏。
正是基于這一背景,我們的三篇論文圍繞單細胞蛋白質組數據分析進行了深入研究。我們首次為單細胞蛋白質組提供了一套系統的 AI 分析方法和數據知識庫。
其中,第一篇論文收集了目前世界上最全面的、不同來源、不同測序技術、不同物種的單細胞蛋白質組數據,并進行了標準化處理和系統性評估。
第二篇論文基于遷移學習技術,從單細胞蛋白質組數據中推斷組織蛋白質組中的細胞比例;
第三篇論文則采用對比學習方法對單細胞蛋白質組進行表征;
我們的計算方法通過實驗驗證,明顯優于直接應用單細胞轉錄組的方法。這些方法已經開源,并配備了詳盡的使用說明,可供全球范圍內的研究人員使用。
我們的算法特別適合那些從事單細胞蛋白質組數據生成的團隊,他們可以直接應用我們的技術進行細胞級別的數據分析和下游應用。
對于臨床醫學專家而言,他們可以利用我們的反卷積算法分析公開的TCGA或CPTAC等蛋白質組數據庫,或者基于自己收集的臨床組織樣本,以深入理解腫瘤微環境,輔助疾病機制的研究和診斷預測。
此外,我們的數據庫允許生物學家和醫學工作者在線探索他們感興趣的蛋白質或細胞類型,觀察這些蛋白質在不同細胞類型中的變化規律,從而支持他們在特定蛋白質研究方向上的研究。
楊帆:在此之前,我們團隊已經進行了大量工作,包括醫學多模態數據分析、疾病預測以及精準醫療等領域的研究。同時,我們也在單細胞轉錄組和空間組學等生命科學基礎計算領域進行了深入探索,并在多個AI頂級會議和期刊上發表了相關論文。
因此,我們在醫學、生命科學、精準醫療和數據分析等領域積累了豐富的經驗。
舉個例子:
我們在預訓練語言模型尚未廣泛應用于單細胞數據分析領域時,就意識到預訓練模型在自然語言處理(NLP)領域已經取得了巨大成功。當時,單細胞數據分析主要依賴于簡單的機器學習方法,并且常常需要針對每個數據集進行手工處理,這限制了模型的泛化能力。
針對這一問題,我們在 2021 年啟動了一個項目,設計了一種基于單細胞數據的大規模預訓練語言模型,名為scBERT。我們根據單細胞數據的特性,開發了基因嵌入(gene embedding)和表達嵌入(expression embedding),使得這些數據能夠被 Transformer 這種先進的計算模型處理和識別。
我們首次引入了 BERT這種預訓練和微調的范式,從而充分利用了當時尚未充分利用的大規模單細胞數據進行預訓練,顯著提升了模型的泛化性和處理跨批次、跨數據集數據的能力。
這一成果發表在了 Nature Machine Intelligence上,開啟了單細胞大模型研究的新篇章。
在這三篇論文發表之后,我們計劃更加聚焦于重大科學問題的研究,并注重其臨床應用和轉化。我們將進一步整合多組學數據和蛋白質大模型,賦能更多的應用場景。
姚建華:我可以補充一些背景信息。
眾所周知,生物體內的核心法則是中心法則,即 DNA、RNA 和蛋白質之間的關系。
DNA 攜帶遺傳信息,通過轉錄成為 RNA,形成轉錄組。
而RNA進一步翻譯成蛋白質,即蛋白質組。
我們的研究工作正是基于這一原理。基因測序技術的發展歷程顯示,DNA 測序是相對容易的部分,而 RNA 和蛋白質的測序難度逐漸增加,因為它們需要更復雜的擴增和測量技術。
從上個世紀 70 年代開始,人類基因組測序技術已經經歷了幾代的發展。
最初,人類主要關注 DNA 信息的測序。大約 10 年前,單細胞技術開始興起,最初主要集中在 RNA 信息的測序。而單細胞蛋白質組學則是最近五六年才開始發展的新興技術。
我們的研究工作也是沿著這一脈絡逐步推進的,從較簡單的數據開始,逐步過渡到更復雜的數據分析。
例如,我們之前的工作 scBERT 主要針對轉錄組數據進行分析。而現在,我們進一步研究蛋白質組數據,這是一個更為復雜和具有挑戰性的領域。隨著數據難度的增加,對算法和計算能力的要求也越來越高。我們的研究正是在這一背景下不斷進步和發展的。
不知道我這樣理解是否正確,請兩位再介紹下三篇論文的聯系與區別。以及,全球范圍內,還有哪些課題組或企業在做類似的工作?
楊帆:您的理解非常準確。
數據資源庫是算法研究的基石,我們深知AI算法的發展離不開數據的支撐。在單細胞蛋白組學領域,數據的準確表征是進行下游應用的關鍵。
掌握了單細胞蛋白組數據后,我們能夠詳細了解每種細胞類型在細胞內蛋白質表達的模式。
基于這些數據,結合AI算法,我們可以進一步推斷組織蛋白組中細胞類型的比例,這對于理解腫瘤微環境至關重要。
目前,臨床上已有大量基于組織蛋白組的數據,這些數據通常來源于腫瘤患者癌組織及其周圍正常組織的樣本,通過質譜技術獲得的是多種細胞類型混合后的蛋白質表達平均水平。
我們的反卷積算法能夠精確推斷出不同細胞類型的比例,使全球研究者能夠從公開數據集中挖掘出有關細胞比例的信息,從而更好地理解腫瘤微環境。
此外,即使在無法進行單細胞蛋白組測序的臨床情況下,我們的算法也能提供一種解決方案,幫助理解細胞微環境,從而輔助臨床進行疾病預后和預測。
這三篇論文可以視為一個整體,其中數據資源庫為基底,上面有兩個不同角度的AI應用,如同一棵大樹上結出的兩個果實。
據我們所知,目前全球范圍內尚無其他團隊或企業開展與我們完全相同的工作。其他機構主要在進行單細胞轉錄組或蛋白質結構的研究,這些研究當然也很重要,但我們的工作填補了單細胞蛋白組學領域的一個空白,具有創新性和前瞻性,未來必將吸引更多研究聚焦于此領域。
姚建華:正如楊帆所提到的,蛋白質結構在AI領域中,尤其是 AlphaFold 這樣的技術最為人所熟知。
AlphaFold 主要分析的是單個蛋白質的結構,例如蛋白質的折疊方式或幾個蛋白質之間的相互作用,它關注的是單個蛋白質的三維結構,以及其功能和對人體細胞的作用。
而我們的研究則是從另一個角度出發,分析細胞內所有蛋白質的表達模式。
我們知道,人體有數以億計的蛋白質,即使是單個細胞內也有成千上萬的蛋白質。我們的目標是分析這些蛋白質之間的相互作用和表達模式,這些信息反映了整個細胞的狀態和微環境。
通過蛋白質組或轉錄組等組學數據,我們可以更全面地理解細胞的微環境和疾病產生的原因,這對于臨床治療和疾病機制的研究具有重要意義。
與 AlphaFold 等關注單個蛋白質結構的技術相比,我們的研究更側重于整個細胞和微環境的系統性分析,這使得我們的工作更接近臨床應用和疾病機制的探索。
楊帆:這三篇論文是在同一個大的研究方向下自然展開的。主要作者包括我和姚老師。
此外,我們的團隊還包括來自不同領域的合作者,如生物信息學和 AI 機器學習領域的專家,以及校企聯合培養的學生。
騰訊 AI Lab 作為一個跨學科的平臺,為跨學科AI應用提供了豐富的土壤。實驗室匯集了 數百位頂尖科學家,這為我們的研究提供了強大的支持。
在 AI Lab,我們有來自生物信息學領域的研究員,他們從生物醫學問題出發,收集數據并定義研究問題。
在模型研發階段,尤其是面對原創性研究中的新問題和挑戰時,我們需要AI技術的創新。在這方面,我們有AI領域世界頂級的科學家與我們合作,共同應對圖模型、可信 AI 以及遷移學習等領域的挑戰。
正是在 AI Lab 這樣一個充滿世界級專家、緊密交流和跨學科合作的環境中,我們才能夠激發出創新的火花,并推動一系列跨學科AI應用研究的發展。
我們的實驗室主任張正友老師和AI醫療首席科學家姚建華博士,分別是 IEEE Fellow 和 AIMBE Fellow,ACM fellow,是世界知名的學術領袖。在他們的指導和把關下,我們的研究員在進行科研和創新時更加自信和從容。
一般來說,我們的項目從啟動到成果發表大約需要一年到一年半的時間。
楊帆:我是清華大學的博士畢業生,在博士期間主要從事臨床組學分析的研究。自2016年起,我開始接觸人工智能領域。博士畢業后,我加入了騰訊隨后在 AI Lab 做研究,至今已近六年。在這里,我相當于又完成了一個 AI 領域的博士學位,進行了廣泛的AI研究。
我感覺自己的知識結構像是“T”字型。
一方面,在組學生物數據分析領域有深入的研究和超過十年的經驗;
另一方面,在AI領域,包括多模態研究、醫學影像、臨床文本數據處理、圖模型、深度學習等多個方面都有所涉獵,并發表了相關論文。
這種“一專多能”的背景使我在跨學科領域,如 AI for Science ,能夠提出獨特的見解和研究方向。
姚建華:我們團隊確實需要這樣的跨學科人才。正如楊帆所提到的,AI Lab 涵蓋了人工智能、機器學習、語音識別、多模態等多個研究方向。我們特別注重生命科學領域的人工智能應用,因此團隊中的許多研究員都具備 AI 和生物學的雙重背景。
只有通過這樣的交叉合作,才能真正推動這一領域的發展。我們也經常與其他專注于人工智能的團隊進行技術上的交流和探討,共同促進科學的進步。
三篇論文逐一追問:好在哪、不足在哪、給誰用
|論文一:《SPDB: a comprehensive resource and knowledgebase for proteomic data at the single-cell resolution》

該論文已入選生物信息學領域數據庫方面專業期刊 Nucleic Acids Research
楊帆:SPDB旨在為不同技術類型的單細胞蛋白組學數據提供一個專門的數據處理框架。
我們通過在統一的環境中對來自不同基礎來源的數據進行標準化處理和分析,使得用戶能夠在一個平臺上對比和探索不同技術來源的數據。
為了確保數據集的獨立性和可靠性,SPDB 并沒有直接整合不同來源的數據集,而是提供了對單個數據集的獨立探索功能,以及對同一蛋白質在不同數據集中的對比探索。
在SPDB數據庫建設的初期,我們面臨的一大挑戰是:如何處理和分析一些我們之前未曾接觸過的數據類型。
例如質譜蛋白質組數據,以及這些原始數據的處理程度和存儲格式的多樣性。
我們通過廣泛閱讀相關文獻,并詳細研究每個數據集的源文獻中關于數據處理的描述,為每個數據集制定了針對性的數據處理步驟,從而確保了數據的準確性和可靠性。
目前,SPDB 的一個不足之處在于:缺乏在線工具供用戶直接使用。未來,我們計劃將研究團隊開發的相關算法集成到SPDB平臺上,以便用戶能夠更方便地使用這些工具。
此外,SPDB 目前還沒有提供蛋白質對應的基因表達信息,即轉錄組數據。因此,我們的后續工作將包括為蛋白質表達提供相應的基因表達數據,以便于用戶進行更全面的對比展示和分析。
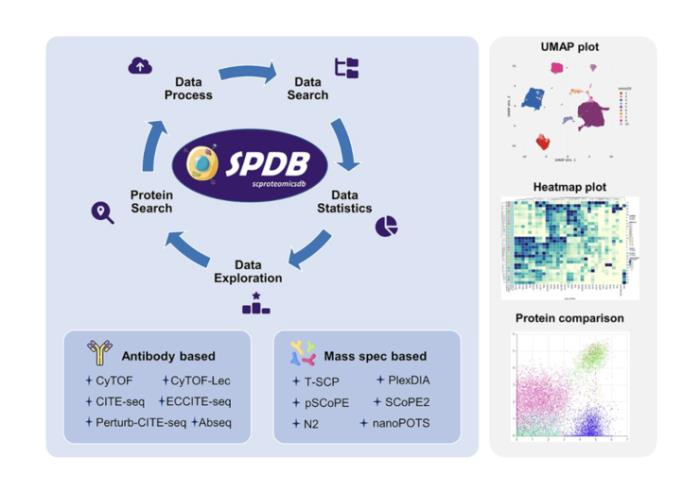
SPDB數據庫 概述圖
楊帆:您的理解非常準確。我們構建這個數據庫的初衷,是因為單細胞轉錄組和空間組學領域的研究已經日益成熟,積累了大量的數據。
市場上也存在一些對單細胞轉錄組和空間組數據進行整合和統計的數據庫,這些數據庫不僅為生物學家和臨床工作者提供了探索和發現的工具,也為生物信息學研究者提供了基于標準化數據進行算法開發的平臺。
由于許多研究者更傾向于使用已經處理好的標準化數據進行開發,而并非所有人都具備從大量分散的原始生物學文獻中提取數據的經驗或知識,我們的論文和工作的目標就是為單細胞蛋白組學領域做出貢獻。
我們希望通過標準化的數據,讓更多的AI研究者和生物信息學工作者能夠看到單細胞蛋白組學數據的潛力,并在此基礎上進行算法的研發和創新。
這就像是為整個單細胞蛋白組學研究社區提供了一片沃土,讓更多創新得以孕育。
此外,這個數據庫也為那些日常工作繁忙、非生物信息學專長的生物科學工作者和醫療工作者提供了便利。有了這個實用的工具,他們可以從單細胞蛋白組學的角度獲得新的啟示和發現,即使這不是他們的主要研究領域。
姚建華:建立這樣一個數據庫的工作量非常巨大,數據分散在各個地方。所以這種工作其實非常適合像我們這樣資源相對充足的公司來開展。
尤其是在大模型時代,數據的重要性愈發凸顯。
以前訓練一個模型可能只需要幾十萬、幾百萬的數據,但現在訓練一個大型模型可能需要數億的數據量。
我們的數據庫已經收集了 3 億個細胞的數據,這樣的數據量才有可能支撐大型模型的訓練。我們將持續更新數據庫,隨著新數據的加入,我們希望這個數據庫能夠真正為整個領域的發展做出貢獻。
|論文二:《 scPROTEIN: a versatile deep graph contrastive learning framework for single-cell proteomics embedding》

已入選 Nature 旗下方法學專業期刊 Nature Methods
為什么要這么做?還有哪些研究不足,應對辦法?
楊帆:scPROTEIN 框架的開發是為了解決單細胞蛋白組數據分析中的獨特挑戰。
在單細胞蛋白組的測定過程中,從細胞分離、裂解、蛋白質提取,到通過質譜技術進行肽段檢測,每一個步驟都可能引入不確定性和噪聲。
例如,樣本制備的差異、標記策略的不同、質譜儀的狀態變化,以及肽段在質譜儀中的離子化和檢測過程,都可能導致批次效應和數據中的噪聲問題。
此外,與單細胞轉錄組數據不同,單細胞蛋白組信號無法通過擴增來增強,只能依靠質譜技術的靈敏度來檢測微量蛋白。
現有的許多單細胞轉錄組數據分析方法,并未充分考慮單細胞蛋白組數據的特殊性,直接應用這些方法效果并不理想。
因此,我們提出了 scPROTEIN 框架,它不僅考慮了單細胞蛋白組數據的層次結構,還采用了基于可信度的方法來估計肽段測定的不確定性,并通過圖對比學習進行表征和去噪,有效解決了數據中的復雜問題。
經過下游任務的充分驗證,scPROTEIN 的性能顯著優于現有的單細胞蛋白組數據分析方法和直接套用單細胞轉錄組的方法。
姚建華:我們的算法實際上提供了一種“數據增強”功能,能夠有效去除數據中的噪聲和批次效應,使得數據分析更為一致和準確。
此外,我們還提出了一種數據編碼的 embedding 方法,這在某種程度上起到了“數據降維”的作用。
正如許多大型模型如 Transformer 和 GPT 所做的那樣,通過 embedding ,我們可以將復雜的蛋白質信息以一種高效的方式表示出來。
這種方法不僅能夠幫助我們提取數據中的核心信息,還能夠揭示不同蛋白質之間的關系,為單細胞蛋白組數據分析提供了一種全新的視角和工具。
楊帆:正如我們之前提到的,scPROTEIN 框架是專門為解決單細胞蛋白組數據所面臨的挑戰而設計的。現有的大多數單細胞數據分析工具,并沒有專門針對單細胞蛋白組數據的特性。例如數據的層次結構和測量不確定性等,進行優化。
scPROTEIN 框架則完全針對單細胞蛋白組數據的特有問題進行了算法開發,因此能夠有效解決這些數據特有的問題。
姚建華:目前而言,幾乎沒有其他方法專門針對單細胞蛋白組分析。這項技術非常前沿,相關數據也相對稀缺,很少有研究能夠收集到如此多的單細胞蛋白組數據。
此外,分析這些數據本身也存在很大的難度,因為數據量大且復雜。
在我們開始這個項目的時候,市場上還沒有專門針對單細胞蛋白組的分析工具,大部分工作都是集中在單細胞轉錄組上。
我們預計在未來幾年,研究者們將會更多地關注蛋白質組學,因此我們在這方面的工作實際上是領先一步,提前進行了探索和開發。
|論文三:《Deep domain adversarial neural network for the deconvolution of cell type mixtures in tissue proteome profiling》

已被Nature旗下機器學習專業期刊 Nature Machine Intelligence 所收錄
能否介紹一下scpDeconv在臨床診斷和治療中的應用前景和潛在挑戰。scpDeconv方法在實際應用中可能遇到哪些問題,以及是否有解決方案。
楊帆:scpDeconv 的臨床應用前景非常廣闊。如我們之前提到的,該方法可以挖掘組織樣本中的細胞比例信息,從而反映腫瘤微環境的狀況。
例如,在我們的研究中,對黑色素瘤樣本進行 scpDeconv 分析后,我們發現不同細胞類型比例的患者預后存在顯著差異。
這種分析可以作為一種輔助診斷工具,幫助醫生預測疾病預后,是精準醫療的一個重要應用場景。
然而,scpDeconv 的潛在挑戰在于:單細胞蛋白質組數據的覆蓋范圍可能不夠廣泛,包括細胞類型和組織類型。
為了克服這一挑戰,我們需要與進行單細胞蛋白質組測序的實驗室合作,共同貢獻更多的公開數據,以便進行更準確的分析。
姚建華:“組織蛋白質組”分析相對容易進行,因為它基于的是整個組織樣本,包括了成千上萬個細胞的蛋白質總和,而“單細胞蛋白質組”分析則需要對每個細胞單獨進行測量,難度和成本都顯著增加。
目前,臨床上主要進行的是組織蛋白質組分析,因為成本較低,技術相對成熟。
我們的scpDeconv 方法,能夠從組織蛋白質組數據中解析出細胞類型的異質性,從而提供類似于單細胞分析的結果,盡管可能不如單細胞數據那么精確,但至少能夠揭示組織中細胞組成的信息。
這樣的技術使得臨床醫生能夠利用現有的數據獲得更多的診斷信息,幫助更準確地進行疾病診斷和治療決策,實現精準醫療的目標。
如何對得起大廠AI lab 的名號?
楊帆:我拋磚引玉,分享一下我們的未來規劃。
首先,我們將貫徹和落實我們實驗室主任張正友博士的指導思想,更加聚焦于解決世界級的重大科學問題,并在 AI for Science 領域實現 AI Lab 的使命——在學術界產生影響,在工業界創造產出。
我們的研究方向與騰訊公司的“科技向善”愿景相契合。未來,我們將繼續利用現有基礎,整合單細胞多組學和蛋白質大模型,推動臨床應用研究,并致力于產出具有世界影響力的原創AI應用研究成果。
姚建華:我們的工作重點是利用人工智能技術解決實際問題和科學挑戰。
作為 AI Lab,我們的優勢在于資源的相對豐富性和研究的聚焦性。與高校相比,公司的環境允許我們集中力量進行大規模的研究項目。
此外,公司的組織結構也使得不同領域的研究員能夠協同合作,共同推進同一項目。雖然高校的研究環境更為自由,但我們這里的研究可以更加集中和深入。
我們的目標是聚焦于最前沿的課題和方向,解決最具挑戰性的問題,以此形成強大的影響力。
我們將繼續在單細胞蛋白質組學領域深耕,不僅推動科學的發展,也為臨床應用提供創新的解決方案。我們期待通過這些努力,為整個領域帶來積極的變化,并為社會做出更大的貢獻。
姚建華:我們目前的重點還是集中在生命科學的一些基礎問題上,如蛋白質和基因組學等領域。
我們確實進行了一些大腦相關的研究,但主要是為了探索大腦的本質。例如,去年我們進行了大腦圖譜的研究,這更偏向于腦科學的基礎研究。
我們試圖通過蛋白質組學和基因組學的信息來區分不同類型的神經元,并理解它們是如何相互聯系和作用的。這樣的研究有助于我們深入理解大腦的機制。
通過我們的AI算法分析基因組學和蛋白質組學數據,我們幫助神經科學家對不同腦細胞進行分類,并描繪它們在大腦中的空間位置。這樣的大腦圖譜研究是神經科學研究的基礎。
當然,要真正深入到腦圖譜的研究,最終還需要回到基因和蛋白質的層面。我們的目標是支持更高層次的科學研究。
姚建華:目前,我們更側重于研究成果的產出,因為工業產出往往需要更多的資源和工程團隊。
我們現階段主要致力于解決一些基礎科學問題。當然,隨著技術積累到一定程度,我們可能會通過與其他團隊合作或尋找合作伙伴來實現這些技術的落地和產業化。
我們的目標是先在科研領域取得突破,為未來的工業應用打下堅實的基礎。
本文作者 吳彤 長期關注人工智能、生命科學和科技一線工作者,習慣系統完整記錄科技的每一次進步。
相關推薦




- 免責聲明
- 本文所包含的觀點僅代表作者個人看法,不代表新火種的觀點。在新火種上獲取的所有信息均不應被視為投資建議。新火種對本文可能提及或鏈接的任何項目不表示認可。 交易和投資涉及高風險,讀者在采取與本文內容相關的任何行動之前,請務必進行充分的盡職調查。最終的決策應該基于您自己的獨立判斷。新火種不對因依賴本文觀點而產生的任何金錢損失負任何責任。
