

 新火種
2023-12-09
新火種
2023-12-09 

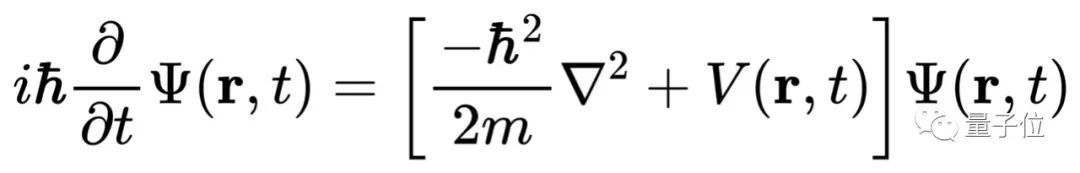

DeepMind開源薛定諤方程求解程序:從量子力學原理出發,TensorFlow實現
只要解出薛定諤方程,你就能預測分子的化學性質。但現實很骨感,迄今為止,科學家只能精確求解一個電子的氫原子,即使是只有兩個電子的氦原子都無能為力。
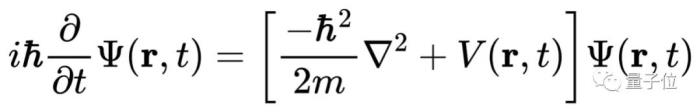
原因是有兩個以上電子組成的薛定諤方程實在太復雜,根本無法精確求解。
既然找不到精確解,科學家們希望能找到一種實用的近似解求法,只要結果足夠近似,也能預測原子或分子的行為。
近日,DeepMind開源了一個“費米網絡”(FermiNet),用來求解分子的電子行為,在30個電子的有機分子上也達到了很高的精度。文章結果發表在期刊Physical Review Research上。
為什么叫費米網絡
在量子力學中,電子沒有精確的位置,我們只能從波函數預測電子在空間中出現的概率,也就是電子云。
比如氫原子的電子云就有以下幾種形態。
曲面內表示電子出現的高概率區域。藍色區域波函數為正,紫色區域波函數為負。(注:波函數平方表示電子出現的概率)
誤差小于0.5%即可預測分子的能量,但這對于化學家來說遠遠不夠,要準確預測分子的形狀和化學性質,需要0.001%的精度,相當于以毫米精度測量足球場寬度。
電子在分子中不僅受到原子核的吸引力、其他電子的斥力,還遵循著量子力學中的費米-狄拉克統計:如果兩個電子交換狀態,波函數要反號。
這也意味著兩個電子的狀態不可能完全相同,否則波函數為0。這在物理中叫做“泡利不相容原理”。
費米網絡正是從這個基本物理原理出發,因此DeepMind將其命名為FermiNet。
交換后符號相反,這可能會讓你想到線性代數中的行列式。行列式任意兩行交換,輸出結果就要乘以-1。
物理學家也是這樣想的,他們用所謂“Slater行列式”來表示電子波函數,但實際情況比Slater行列式要復雜得多,為了更精確表示電子行為,往往需要幾百萬個Slater行列式的線性組合。
工作原理
與函數線性組合相比,神經網絡在表示復雜函數時往往更具有優勢。
在構造FermiNet之初,研究人員就把泡利不相容原理作為第一性原理引入神經網絡。
在FermiNet中,每個電子都有單獨的信息流。不僅如此,他們將網絡每一層所有流平均化,然后傳遞給下一層的每一流。這樣,這些流就具有正確的反對稱性要求。
而且在FermiNet行列式中的每個元素都包含所有電子,效率遠遠超出波函數只有單個電子的情況。
與Slater行列式不同,FermiNet是通用函數逼近器,如果神經網絡層變得足夠寬,則可以無限逼近真實波函數。
這意味著,如果我們正確地訓練這些網絡,它們應該能夠將幾乎完全精確的解擬合到薛定諤方程。
訓練是通過最小化系統的能量來擬合FermiNet。FermiNet用蒙特卡洛方法隨機選擇電子構型,在每個電子排列中局部評估能量,累加每個排列的貢獻,并將其最小化。
實驗結果
研究人員將FermiNet用在具有10個電子以內的原子上,能量精度均在99.8%左右。
對于30個電子的環二丁烷,FermiNet算出的能量達到了97%的精度,雖然精度不是很高,但DeepMind表示,作為一種“便宜但不夠準確”的方法,這是巨大的成就。
現在FermiNet已經在GitHub上開源,代碼基于TensorFlow實現,如果你想用它算一算氫分子,不妨試試這串代碼:
import sys
from absl import loggingfrom ferminet.utils import systemfrom ferminet import train
# Optional, for also printing training progress to STDOUTlogging.get_absl_handler().python_handler.stream = sys.stdoutlogging.set_verbosity(logging.INFO)
# Define H2 moleculemolecule = [system.Atom('H', (0, 0, -1)), system.Atom('H', (0, 0, 1))]
train.train(molecule=molecule,spins=(1, 1),batch_size=256,pretrain_config=train.PretrainConfig(iterations=100),logging_config=train.LoggingConfig(result_path='H2'
經過100次迭代后,該程序會輸出一個氫原子波函數文件。官方建議最好用GPU來運行,因為他們計算乙烯分子就用8個GPU花2天時間才算出。
除了研究電子外,DeepMind還將神經網絡用于其他基礎科學研究,比如蛋白質折疊、玻璃態動力學、晶格量子色動力學等。
Tags:
相關推薦




- 免責聲明
- 本文所包含的觀點僅代表作者個人看法,不代表新火種的觀點。在新火種上獲取的所有信息均不應被視為投資建議。新火種對本文可能提及或鏈接的任何項目不表示認可。 交易和投資涉及高風險,讀者在采取與本文內容相關的任何行動之前,請務必進行充分的盡職調查。最終的決策應該基于您自己的獨立判斷。新火種不對因依賴本文觀點而產生的任何金錢損失負任何責任。
