

 新火種
2024-12-24
新火種
2024-12-24 


麥吉爾大學DingLab基于深度學習開發單細胞水平轉座子位點表達定量模型,登Nature子刊

編輯丨ScineceAI
該論文介紹 MATES:一種基于深度學習的單細胞水平轉座子定量工具。MATES 使用基于自編碼器的模型,通過分析轉座子區域周圍獨特比對讀段的分布,概率性地將多重比對轉座子讀段分配到特定位點。通過深度神經網絡,MATES 學習獨特讀段分布與多重比對讀段來自特定位點的可能性之間的關系。
這一創新方法在多個單細胞測序平臺上進行了嚴格驗證,包括 10X Genomics(scRNA-seq、scATAC-seq 和 Multiome)、Smart-seq 和空間轉錄組學(10X Visium)。該工具的多樣性和精確性已帶來了新的生物學見解,為更廣泛的應用和實驗驗證鋪平了道路。
該研究以「MATES: a deep learning-based model for locus-specific quantification of transposable elements in single cell」為題,于 2024 年 10 月 11 日發布在《Nature Communications》。

背景介紹
轉座子(Transposon),又稱轉座元件或跳躍基因,是哺乳動物基因組的重要組成部分,在基因調控、基因組進化和細胞間異質性中發揮著關鍵作用。盡管部分轉座子仍然活躍并能夠在基因組中跳躍,但大多數轉座子已經積累了突變和退化,使其失去了主動轉座的能力。因此,許多轉座子被保留在基因組中并作為調控元件發揮作用。這些非編碼功能包括調控基因表達以及形成長鏈非編碼 RNA(lncRNA),這些 RNA 參與關鍵的調控網絡,影響基因表達和細胞功能。盡管轉座子扮演著這些重要角色,但由于其重復序列和高拷貝數導致的多重比對測序讀段(reads)定量的挑戰,我們在單細胞水平上對特定位點轉座子的理解仍然有限。
轉座子高多重比對讀段的挑戰
從轉座子的高對比讀段中準確量化表達量是這個領域的一項重大挑戰。許多現有的單細胞轉座子量化工具過度依賴比對算法來處理多重比對讀段。然而由于轉座子的重復性,利用對比算法量化轉座子表達量存在局限性,即他們忽略了轉座子周圍的基因組上下文信息。
特定位點轉座子量化的挑戰
現有的方法未能實現精確的特定位點轉座子定量,他們有些只量化轉座子亞家族的表達量,有的僅僅將讀段分配到對比算法提供的“最佳” 位置,因此在處理轉座子區域中普遍存在的多重比對讀段方面非常有限。這些方法忽視或回避了由轉座子重復特性導致的多重比對讀段分配挑戰,這種忽視可能低估了轉座子定量中分配多重比對讀段的復雜性和不確定性。
方法總結
利用深度學習模型解決轉座子多重比對讀段的挑戰
為了解決這些挑戰并填補空白,我們提出了 MATES,這是一種基于深度神經網絡的方法,專為跨模式的單細胞測序數據中的特定位點轉座子精確定量而設計。MATES 利用轉座子位點周圍獨特比對讀段的分布信息,將多重比對轉座子讀段分配到特定位點,從而實現特定位點轉座子的定量。通過深度神經網絡的強大能力,MATES 捕捉轉座子位點周圍獨特比對讀段分布與多重比對讀段被分配到這些位點的概率之間的復雜關系。該方法使 MATES 能夠基于轉座子位點的局部上下文,以概率方式處理多重比對讀段的分配問題。
MATES 方法包括多個關鍵步驟。首先,將原始讀段比對到參考基因組,識別唯一比對到某個轉座子位點的讀段(獨特比對讀段)以及比對到多個轉座子位點的讀段(多重比對讀段)。接著,為每個轉座子位點計算覆蓋向量,表示圍繞該位點的獨特比對讀段分布(上下文),并將每個轉座子區域細分為長度為 W(如 10 個堿基對)的較小區間。根據區間內獨特比對讀段和多重比對讀段的比例,將每個區間分類為獨特占主導區域或多重占主導區域。然后,MATES 使用自編碼器模型學習潛在嵌入,用于表示轉座子位點的高維獨特讀段覆蓋向量,即特定位點的比對上下文。
此外,轉座子家族的獨熱編碼信息也作為模型輸入,用于結合潛在嵌入預測特定位點的多重比對比例 (α)。模型的總損失由重構損失和相鄰區間讀段覆蓋連續性的損失組成,后者反映了多重占主導區間的覆蓋應接近其相鄰的獨特占主導區間。通過訓練完成的模型,我們可以以概率方式統計每個轉座子位點的讀段總數,從而實現轉座子在位點水平的精確定量。進一步地,通過將轉座子定量與單細胞數據中的傳統基因定量(如基因表達或基因可及性)結合,MATES 能夠更準確地對細胞進行聚類,并識別全面的生物標志物(基因和轉座子),以表征獲得的細胞群。除了高效處理單細胞數據的各種模式,MATES 還提供特定位點轉座子的可視化功能,支持生成 bigwig 文件和交互式基因組查看器(IGV)圖,幫助研究人員直觀地探索和解釋基因組中轉座子位點的讀段分配。
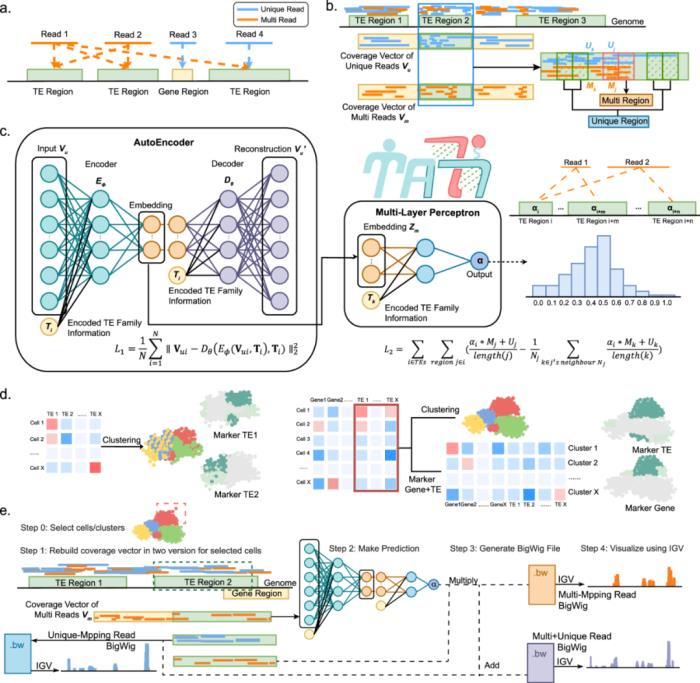
圖1:MATES方法概覽。
a原始讀段被比對到參考基因組,同時考慮轉座子位點上的多重比對讀段。b 構建轉座子覆蓋向量,包括獨特比對讀段覆蓋向量 V_u 和多重比對讀段覆蓋向量 V_m,以捕獲讀段的分布信息。c 自編碼器(AutoEncoder)模型從獨特比對讀段覆蓋向量中提取潛在嵌入。這些嵌入與轉座子家族數據 T_i 結合,用于預測多重比對讀段與每個轉座子位點匹配的概率α。d MATES 計算的多重比對概率 α 對構建轉座子計數矩陣至關重要。該矩陣是細胞分析的關鍵,可單獨使用,也可與傳統基因計數矩陣結合使用。結合使用可以增強細胞聚類和生物標志物(基因和轉座子)的發現,從而更全面地理解細胞特性。e MATES 實現基因組范圍的讀段覆蓋可視化,并生成基于基因組瀏覽器的可視化文件。該方法在單個細胞中對特定位點的 轉座子進行定量,生成包含從概率分配的多重比對讀段計算的覆蓋的 bigwig 文件。這些文件將獨特和多重比對讀段的覆蓋信息合并,生成全面的 bigwig 文件,可通過交互式基因組查看器(IGV)等工具實現基因組范圍的轉座子讀段可視化。
結果展示
在我們對 MATES 的系統性評估中,使用了不同測序平臺、模式和物種的多種單細胞數據集,結果表明 MATES 始終能夠提供更準確的轉座子定量結果。除了更高的精確性之外,MATES 還提供了特定位點水平的轉座子定量,并且能夠在不同測序平臺和數據模式下推廣使用,從而更全面地理解轉座子在細胞動態和基因調控中的作用。我們還通過 Nanopore 和 PacBio 長讀段測序以及模擬數據對方法的預測結果進行了驗證。通過將 MATES 的單細胞轉座子定量與模擬的真實值或長讀段測序的代理真實值進行比較,我們證明了 MATES 的準確性及其相較于現有方法的優勢。結果表明,MATES 在探索轉座子在單細胞生物學中的作用方面表現出色,并為不同實驗背景下的轉座子定量提供了一個實用的解決方案。
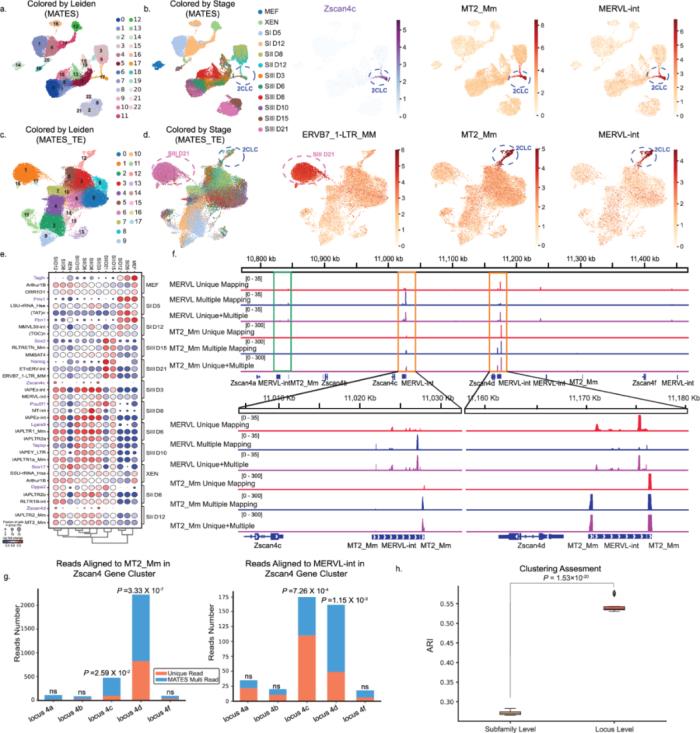
圖2: MATES 增強小鼠化學重編程(10X scRNA-seq數據)中的細胞聚類與生物標志物發現。
a, b UMAP 圖顯示通過整合轉座子和基因,MATES 在細胞聚類中的效果。a 圖按 Leiden 聚類結果著色,而 b 圖按重編程階段著色,突出顯示了鑒定的基因(紫色)和 轉座子(紅色)生物標志物。c, d 額外的 UMAP 圖強調僅使用轉座子進行聚類的 MATES 能力,c 圖按 Leiden 聚類著色,d 圖按重編程階段著色。值得注意的是,MT2_Mm 和 MERVL-int 轉座子是 Zscan4c/Zscan4d 陽性細胞中的重要標志物,與已知的 2CLCs 標志一致。e點圖展示 MATES 鑒定的階段特異性標志基因(紫色)和轉座子黑色)。f 示意圖展示 MATES 的概率性方法如何將多重比對讀段分配到特定轉座子位點,特別是在 2CLCs 中與 Zscan4c/Zscan4d 位點相關的 MT2_Mm 和 MERVL-int。g 條形圖顯示 MT2_Mm 和 MERVL-int 在 Zscan4c/Zscan4d 位點的讀段富集情況。富集的 p 值通過單側二項檢驗計算。h箱線圖比較 MATES 在特定位點水平和亞家族水平轉座子定量下的細胞聚類效率,使用調整蘭德指數(ARI)評估。
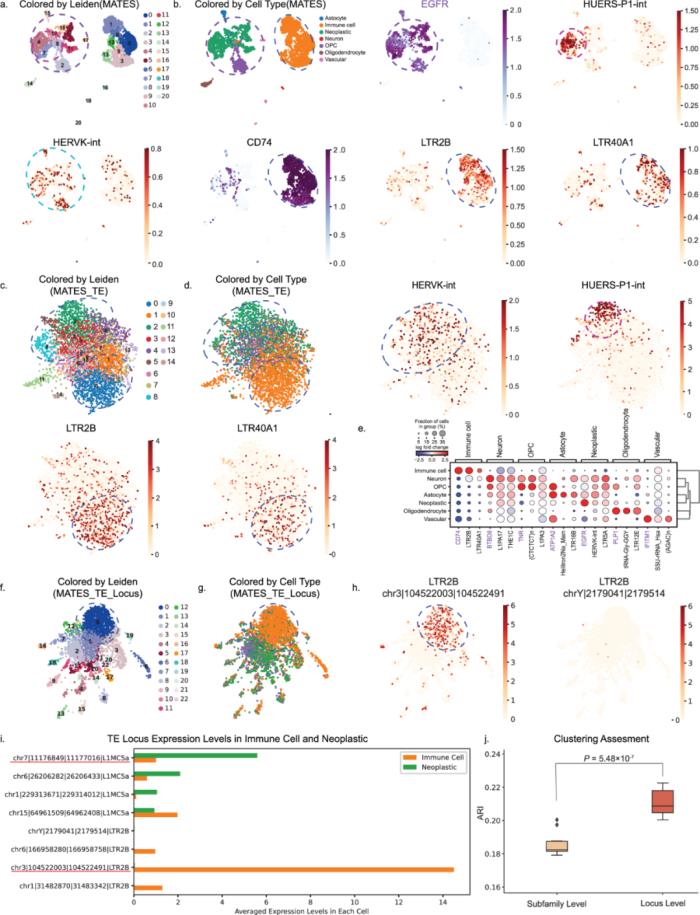
圖 3:MATES 在 Smart-Seq2 單細胞 RNA-seq 數據中定量與疾病相關的轉座子表達。
a, b UMAP 圖展示了基于基因和轉座子標志物的細胞聚類。MATES 或 Gene+TE 表示通過 MATES 定量的基因表達與轉座子數據相結合。UMAP 初始按 Leiden 聚類著色(a),隨后按細胞類型及特異標志物著色,包括腫瘤標志(EGFR、HUERS-P1-int 和 HERVK-int)及免疫細胞標志(CD74、LTR2B 和 LTR40A1)(b)。c, d 基于 MATES 定量的轉座子表達生成的 UMAP 圖,分別按 Leiden 聚類(c)和按標志物(如 HERVK-int)著色的細胞類型(d)。e 點圖揭示了 MATES 鑒定的標志基因、轉座子和細胞類型之間的關聯。f, g 展示了利用 MATES 的特定位點轉座子定量增強聚類精確性的結果,f 圖顯示 Leiden 聚類,g 圖顯示細胞類型。h 列出免疫細胞中特定位點水平高表達的轉座子標志物(LTR2B)及其非表達位點,證明了 MATES 在位點水平轉座子定量方面的能力。i 條形圖展示免疫細胞和腫瘤細胞中轉座子的平均特定位點表達水平。j 箱線圖比較了基于 MATES 的特定位點水平和亞家族水平轉座子定量的細胞聚類效果,通過調整蘭德指數(ARI)評估,表明 MATES 在生物標志物識別和細胞分類中的分辨率提升。
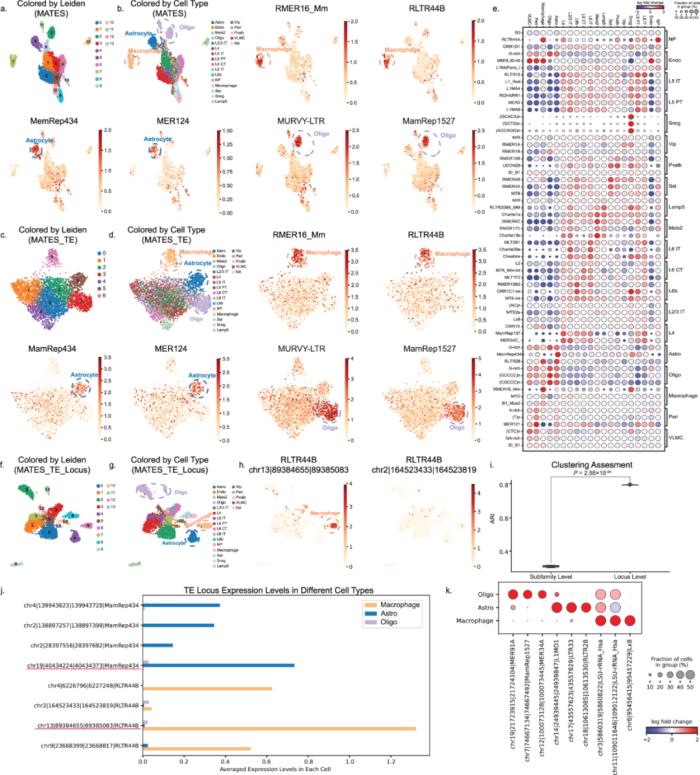
圖 4:MATES 在成年小鼠大腦 scATAC-seq 數據中的多樣性應用。
a–d UMAP 圖展示了 MATES 在細胞聚類和識別特征性轉座子標志物中的定量效果,結合轉座子和峰值數據進行聚類分析。a 圖顯示 Leiden 聚類結果,b 圖按細胞類型及轉座子標志物著色,鑒定出了關鍵轉座子標志物,例如在巨噬細胞中的 RMER16_Mm 和 RLTR44B,在星形膠質細胞中的 MamRep434 和 MER124,以及在少突膠質細胞中的 MURVY-LTR 和 MamRep1527。c 和 d 圖展示了 MATES 在以轉座子為中心的聚類中的特異性,僅使用 MATES 的轉座子定量數據進行聚類分析,其中 c 圖聚焦于 Leiden 聚類,d 圖展示細胞類型及之前提到的特征性轉座子標志物。e 點圖簡明呈現了 MATES 鑒定的細胞類型特異性轉座子標志物。f–h 這些面板說明了 MATES 使用特定位點水平轉座子定量提升聚類準確性的效果。f 圖展示了基于特定位點水平轉座子定量的 UMAP 可視化,按顏色標識 Leiden 聚類;g 圖展示了相同的 UMAP,但按顏色區分不同細胞類型;h 圖提供了特定位點水平的轉座子標志物 RLTR44B 在巨噬細胞中的具體示例,并與該轉座子一個未開放位點進行對比,體現了 MATES 在詳細特定位點水平轉座子定量方面的能力。i 箱線圖比較了 MATES 在特定位點水平與亞家族水平轉座子定量下的細胞聚類效率(調整蘭德指數,ARI),突出了采用特定位點轉座子定量的優勢。j 條形圖展示巨噬細胞、少突膠質細胞和星形膠質細胞中特定位點轉座子的平均表達水平。k 點圖顯示了 MATES 鑒定的特定位點水平轉座子標志物及其對應的細胞類型。
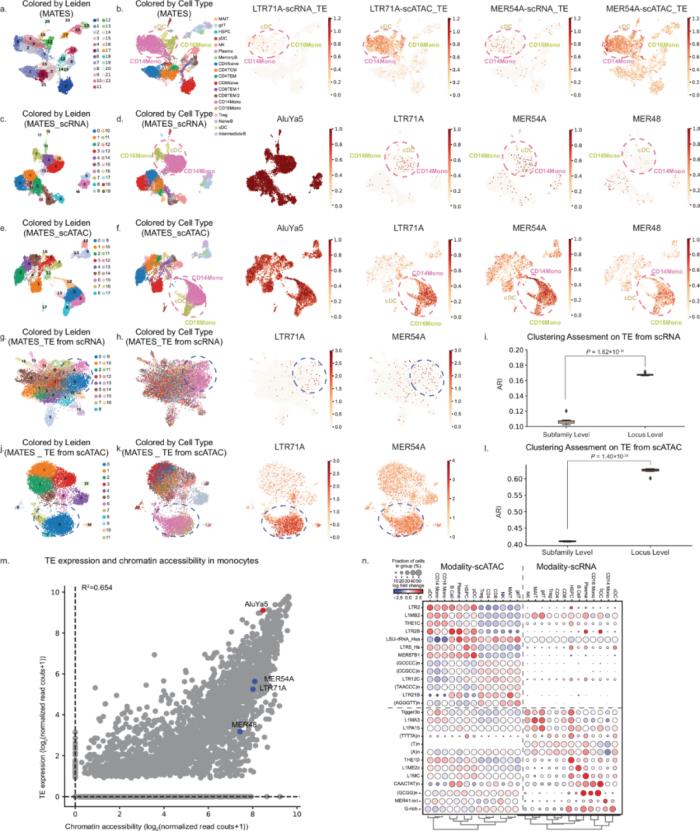
圖 5:使用 MATES 對人類 PBMCs 進行多模態轉座子分析。
a, b UMAP 圖展示了通過整合scRNA和 scATAC 模態的 MATES 聚類結果,a 圖顯示 Leiden 聚類,b 圖展示細胞類型聚類。c–f 跨模態的轉座子定量突出多模態定量的互補性。c 和 d 圖展示了在 scRNA 模態中基因和轉座子聯合的 UMAP 聚類,e 和 f 圖則展示了在 scATAC 模態中峰值和轉座子聯合的 UMAP 聚類。c 和 e 按 Leiden 聚類著色,d 和 f 按細胞類型著色,展示了轉座子如 AluYa5 在兩種模態中的差異性表達,而 MER48、LTR71A 和 MER54A 則特異于 scATAC 模態。g–l 這一系列 UMAP 圖和箱線圖說明了多模態轉座子分析。g 和 j 是轉座子表達的 UMAP 圖,按 Leiden 聚類著色以突出聚類模式;h 和 k 是聚焦于不同細胞類型及其轉座子標志物的 UMAP 圖,提供了細胞特性和相關轉座子的深入見解;i 和 l 是比較細胞聚類效果的箱線圖(調整蘭德指數,ARI),強調特定位點水平定量相比亞家族水平定量提供的更高分辨率。m 展示了通過 scRNA 和 scATAC 模態鑒定的轉座子標志物,指出高表達的轉座子通常與染色質可及性增強相關,而反向情況并非普遍觀察到,突顯了每種模態的獨特貢獻。n 點圖捕捉了每種細胞類型的特征性轉座子,驗證了 scATAC 和 scRNA 數據的互補性,為全面了解轉座子動態提供了支持。
總結與展望
MATES 利用自編碼器,基于獨特占主導地位的轉座子區域的覆蓋向量,學習單個轉座子位點上獨特比對讀段的分布模式。它整合了獨特比對和多重比對讀段,從而在位點水平上精確定量轉座子表達。該工具不僅限于亞家族級別的轉座子表達定量,還能夠實現位點水平的定量,從而提高細胞群體分析的分辨率,并推動特定位點轉座子標志物的識別。
論文鏈接:https://www.nature.com/articles/s41467-024-53114-7
相關推薦




- 免責聲明
- 本文所包含的觀點僅代表作者個人看法,不代表新火種的觀點。在新火種上獲取的所有信息均不應被視為投資建議。新火種對本文可能提及或鏈接的任何項目不表示認可。 交易和投資涉及高風險,讀者在采取與本文內容相關的任何行動之前,請務必進行充分的盡職調查。最終的決策應該基于您自己的獨立判斷。新火種不對因依賴本文觀點而產生的任何金錢損失負任何責任。
