

 新火種
2024-12-05
新火種
2024-12-05 


僅總參數量0.1%、單GPU15分鐘完成微調,人類基因組基礎模型NT登Nature子刊

編輯 | 蘿卜皮
從 DNA 序列預測分子表型仍然是基因組學中的一個長期挑戰,通常是由于注釋數據有限以及無法在任務之間轉移學習所致。
在這里,英國倫敦InstaDeep的研究人員提出了在 DNA 序列上進行預訓練的基礎模型,稱為 Nucleotide Transformer;其參數范圍從 5000 萬到 25 億,并整合了來自 3,202 個人類基因組和 850 個不同物種基因組的信息。
這些 Transformer 模型可生成特定上下文的核苷酸序列表示,即使在低數據環境下也能實現準確預測。該模型可以以低成本進行微調,來解決各種基因組學應用問題,為從 DNA 序列進行準確的分子表型預測提供了一種廣泛適用的方法。
該研究以「Nucleotide Transformer: building and evaluating robust foundation models for human genomics」為題,于 2024 年 11 月 28 日發布在《Nature Methods》。

基礎人工智能(AI)模型通過大規模訓練,能夠處理各種預測任務,顯著推動了科技領域的發展。自然語言處理(NLP)中的掩碼語言建模,是實現語言理解的一種方法,通過預測句子中的缺失單詞,訓練模型深入理解語言。
生物學中的早期基礎模型通過訓練蛋白質序列,利用掩碼氨基酸預測任務,展示了在蛋白質結構和功能預測等任務中的優越表現。遷移學習使得這些蛋白質語言模型在數據稀缺情況下超越了傳統方法。
除了蛋白質序列,DNA 序列中的編碼模式在理解基因組過程中的重要性日益突出,特別是在評估變異影響等方面。現代基因組學面臨著數據量龐大、數據模式異常復雜的挑戰。
在這里,英國倫敦 InstaDeep 構建了強大的基礎模型來編碼基因組序列,稱為 Nucleotide Transformer(NT),并提出了系統的研究和基準來評估它們的性能。
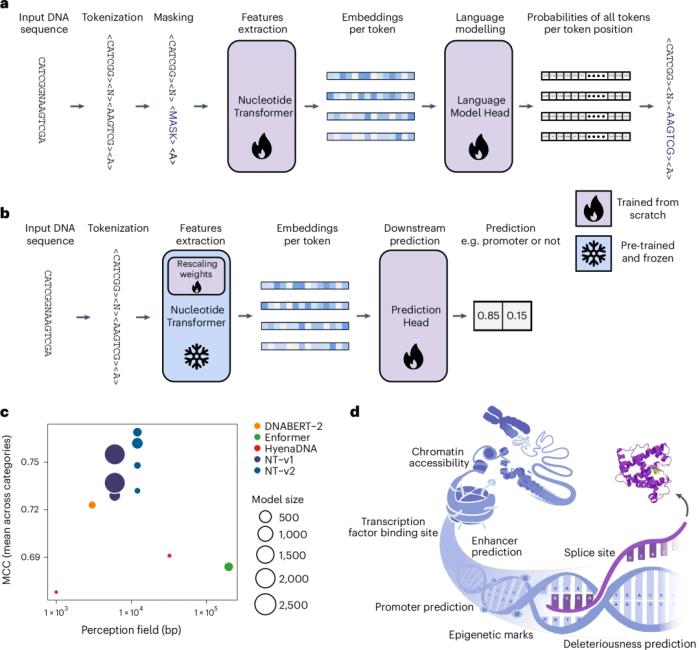
該團隊通過構建四個大小各異的不同 LM 來進行他們的研究,這些 LM 的參數數量從 5 億到 25 億不等,比 DNABERT-2 大 20 倍,比 Enformer 主干模型大 10 倍。這些模型在三個不同的數據集上進行了預訓練,包括人類參考基因組、3,202 個不同人類基因組的集合以及來自不同物種的 850 個基因組。
為了評估 NT 在適應各種任務時的性能穩定性,團隊在 18 個不同的基因組策劃預測任務上訓練了每個模型,并使用系統的十倍交叉驗證程序將它們與三個替代 DNA 基礎模型以及一個最先進的非基礎模型進行了比較。
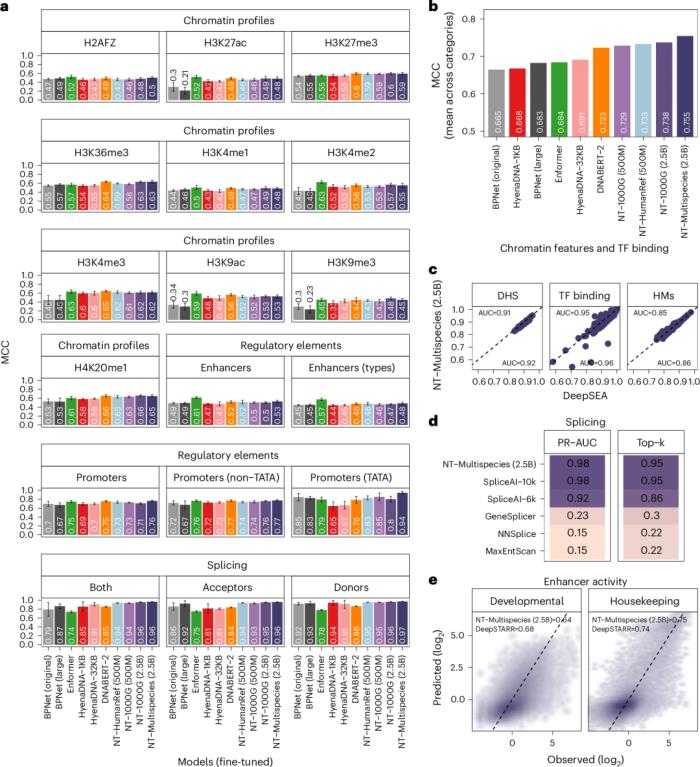
圖示:Nucleotide Transformer 模型在微調后準確預測了不同的基因組學任務。(來源:論文)
此外,為了擴大評估范圍,研究人員將表現最佳的模型與三個針對特定任務進行了優化的最先進的監督基線模型進行了比較。
為了解讀在預訓練期間學習到的序列特征,團隊探索了模型的注意力圖和困惑度,并對它們的嵌入進行了數據降維。
此外,研究人員通過基于零樣本的評分評估了嵌入對人類功能重要遺傳變異影響的建模能力。在初始實驗結果的基礎上,他們開發了第二組四個 LM,其參數大小從 5 億個減少到 5000 萬個,以研究此類模型的縮放規律。
該團隊成功構建了一個模型,僅使用十分之一的參數數量、將感知場大小增加了一倍,便達到了之前最佳模型的性能。
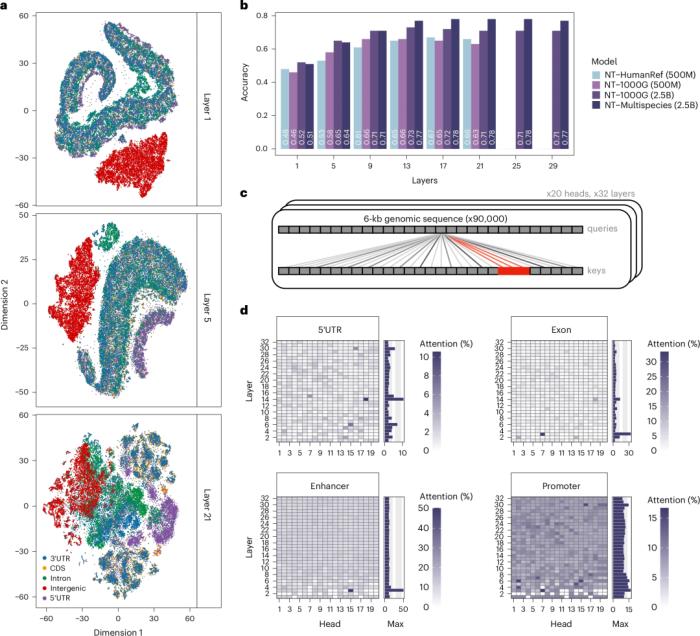
圖示:Nucleotide Transformer 模型獲得了有關基因組元件的知識。(來源:論文)
并且,與全模型微調 (IA3) 相比,該研究提供了相對快速且資源高效的微調程序,且差異很小。
值得注意的是,該團隊的微調方法僅需要總參數數量的 0.1%,即使是這里最大的模型也可以在單個 GPU 上在 15 分鐘內完成微調。
與廣泛的探索練習相比,這種技術在使用更少的計算資源的情況下產生了更好的結果,證實了下游模型工程可以帶來性能大幅改進和優化。
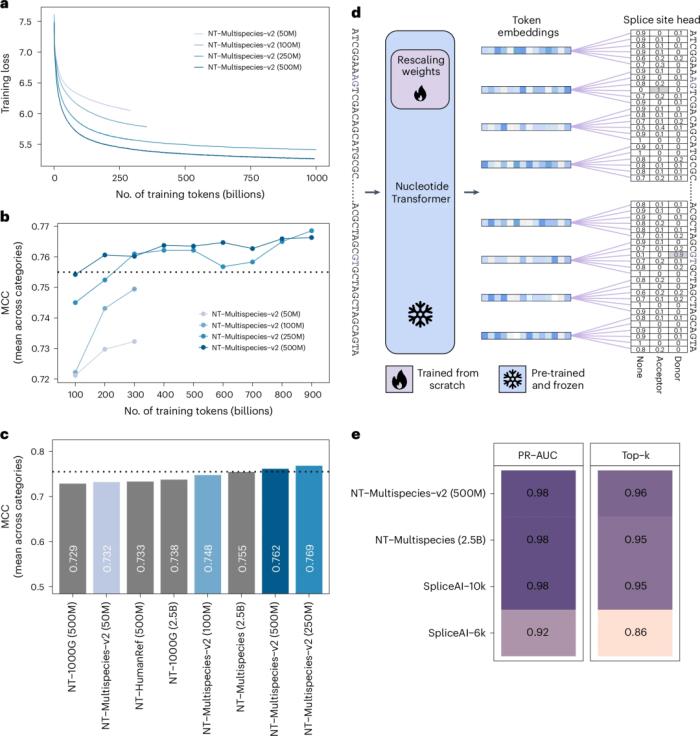
圖示:高效的模型架構允許匹配性能,同時大幅減少模型參數的數量。(來源:論文)
總之,該研究的結果基于不同的基因組預測任務,表明物種內(在單個物種的多個基因組上進行訓練時)和物種間(在不同物種的基因組上進行訓練時)的變異性都會顯著影響任務間的準確性。
在大多數考慮的人類預測任務中,用不同物種的基因組訓練的模型比只用人類序列訓練的模型表現更好。這表明,用不同物種訓練的 Transformer 模型已經學會了捕捉可能對不同物種具有功能重要性的基因組特征,從而能夠在各種基于人類的預測任務中更好地推廣。
基于這一發現,研究人員認為未來的研究可能會受益于利用跨物種遺傳變異,包括確定對這種變異進行采樣的最佳方式。另一個有趣的途徑是探索對物種內變異進行編碼的不同方式。
同時研究人員表示,這里將所有個體基因組的序列混合在一起的方法只取得了有限的改進,因此表明當大多數基因組是共享的時,利用來自不同個體的基因組可能并不那么簡單。
論文鏈接:
相關推薦




- 免責聲明
- 本文所包含的觀點僅代表作者個人看法,不代表新火種的觀點。在新火種上獲取的所有信息均不應被視為投資建議。新火種對本文可能提及或鏈接的任何項目不表示認可。 交易和投資涉及高風險,讀者在采取與本文內容相關的任何行動之前,請務必進行充分的盡職調查。最終的決策應該基于您自己的獨立判斷。新火種不對因依賴本文觀點而產生的任何金錢損失負任何責任。
