

 新火種
2025-01-09
新火種
2025-01-09 


準確預測蛋白質功能新SOTA,中南大學推出全新深度學習模型,登Nature子刊

編輯丨&
預測蛋白質功能的計算方法對于理解生物學機制和治療復雜疾病具有重要意義。然而,現有的預測計算方法缺乏可解釋性,難以理解蛋白質結構和功能之間的關系。
在研究中,來自中南大學的團隊提出了一種基于深度學習的解決方案,名為 DPFunc,用于使用域引導的結構信息進行準確的蛋白質功能預測。
DPFunc 可以在結構域信息的指導下檢測蛋白質結構中的重要區域并準確預測相應的功能。它優于當前最先進的方法,并與現有的基于結構的方法相比取得了顯著改進。
他們的研究成果以「DPFunc: accurately predicting protein function via deep learning with domain-guided structure information」為題,于 2025 年 1 月 2 日刊登在《Nature Communications》。

詳細分析表明,結構域信息的引導有助于 DPFunc 進行蛋白質功能預測,能夠檢測蛋白質結構中與其功能密切相關的關鍵殘基或區域。故而,該方法是大規模蛋白質功能預測的有效工具。
DPFunc 概述
DPFunc 是一種基于深度學習的方法,用于使用域引導結構信息進行蛋白質功能預測。
它由三個模塊組成:基于預先訓練的蛋白質語言模型和圖神經網絡的殘基級特征學習模塊;蛋白質水平特征學習模塊;蛋白質功能預測模塊。
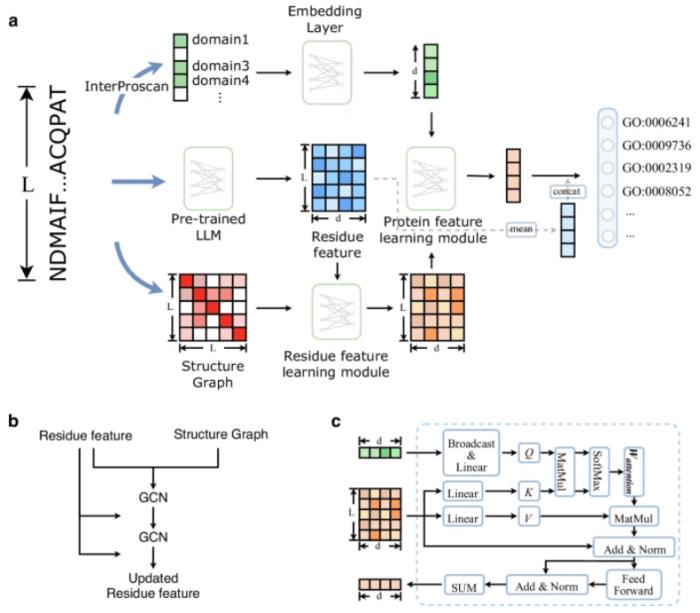
圖 1:DPFunc 的模型架構。(圖源:論文)
殘基水平特征學習模塊將蛋白質序列和結構作為輸入。它首先從預訓練的蛋白質語言模型(ESM-1b)中為每個殘基生成初始特征,根據相應的蛋白質結構構建接觸圖。隨后,這些接觸圖和殘基層特征被進一步饋送到幾個圖神經網絡(GCN)層中,以更新和學習最終的殘基層特征。
為了評估不同殘基的重要性,受 transformer 結構的啟發,團隊引入了一種注意力機制,將蛋白質水平的結構域特征和殘基水平特征交織在一起,從而檢測每個殘基的重要性。
預測結果通過通用的后處理程序進行處理,以確保與基因本體論(GO)項結構的一致性。
為了獲得模型的性能,團隊將其與與僅基于序列和兩種基于結構的方法進行公平比較。他們采用了以前使用過的數據集,在其他模型平均得分近似的情況下,新模型的性能超過了現有模型一大截。這一發現表明,蛋白質序列中包含的結構域信息為蛋白質功能預測提供了有價值的見解。
模型性能分析
團隊根據不同的時間戳將大規模數據集劃分為訓練集、驗證集和測試集。與以前使用的 PDB 數據集不同,這個大規模數據集包含更多的蛋白質和相應的附加信息。
為了確保公平的比較,標準化的方式被應用于后續的所有處理過程。
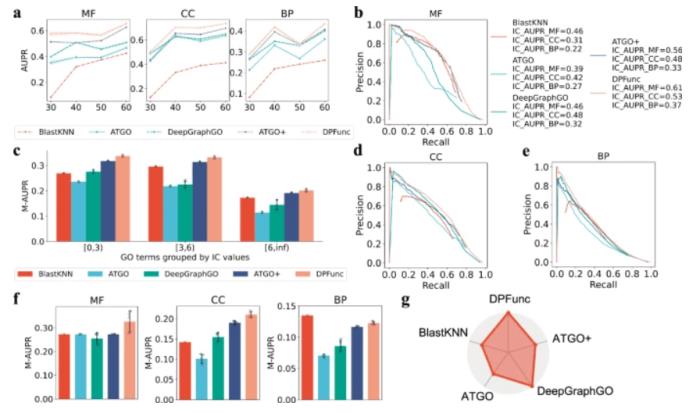
圖 2:模型性能的詳細分析(圖源:論文)
除了整體性能之外,DPFunc 還擅長預測具有高 IC 值特征的信息性 GO 項。由于這些項出現次數少且訓練樣本有限,因此帶來了更大的挑戰。在預測樣本較少的 GO 項時,DPFunc 的性能始終優于其他方法。
DPFunc 表現出優于 SOTA 方法的明顯優勢,特別是它能夠處理具有低序列同一性的不可見蛋白質、具有高 IC 值的信息性 GO 項以及具有更深節點的特定 GO 項。
為了明確證明域信息在 DPFunc 中的關鍵作用,團隊采用平均池化層替換了域注意力塊。憑借領域洞察力,DPFunc 的幾個模塊中的 AUPR 中位數分別提高了 12.0%、14.7% 和 16.3%。這些結果明確證實了整合結構域信息進行蛋白質功能預測的無與倫比的價值。
為了進一步說明 DPFunc 在檢測相似結構基序方面的潛力,即使沒有序列相似性,團隊也進行了兩個案例研究。對兩種將細胞與外部環境分離的關鍵質膜蛋白,DPFunc 能夠捕捉結構相似性并準確預測功能,即使面對不同的序列也是如此,突顯了它在蛋白質功能預測方面的巨大潛力。
除此之外,DPFunc 可有效檢測酶功能的重要活性位點。這種非凡的能力歸功于圖神經網絡的強大功能,它可以聚合來自兩個相鄰活動站點的信息。不過,盡管 DPFunc 有效地進行了檢測,但在無序區域尋找活性位點仍然是一個挑戰,可能會在未來的模型中進一步探索。
學習模塊
團隊使用兩個數據集來評估其方法的性能。前者是一個非冗余集,通過以 95% 的序列同一性對所有 PDB 鏈進行聚類。后者是從 UniProt 和 Gene Ontology 數據庫中收集的。
DPFunc 整合了這兩個模塊并預測了蛋白質功能。具體來說,它利用初始殘基特征和蛋白質特征來注釋功能。此外,一旦該模型經過訓練,它就可以根據注意力機制檢測結構中的重要殘留物。
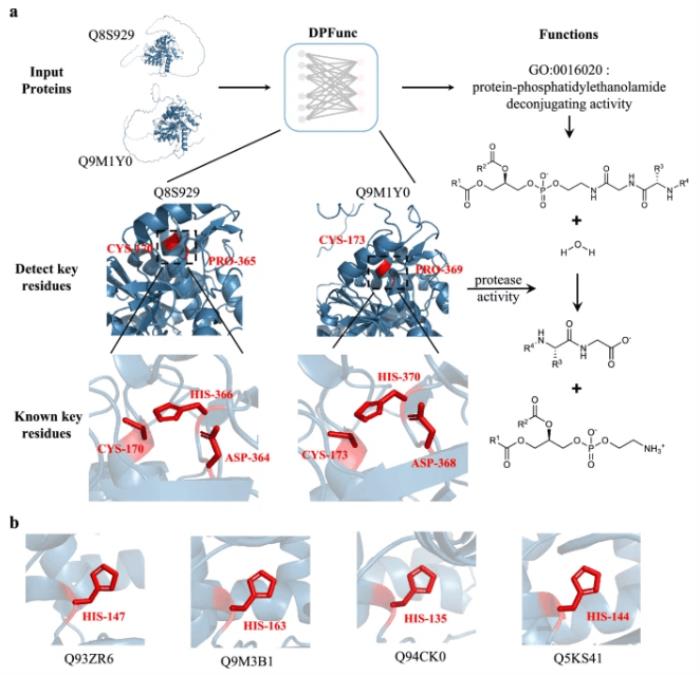
圖 3:DPFunc 檢測到的關鍵殘基。(圖源:論文)
小結
結合了結構域引導的結構信息來識別蛋白質結構中的關鍵區域,從而能夠根據潛在結構基序和關鍵殘基準確預測功能。與其他最先進的深度學習方法的全面比較證明了此次提出的方法的優勢。
DPFunc 在稀有功能、特定功能和與已知蛋白質序列相似性較低的困難蛋白質方面也優于其他方法。其表現出區分不同結構之間蛋白質的令人印象深刻的能力。DPFunc 可以學習相似的結構基序,即使它們的序列相似性并不如訓練集那么高。
DPFunc 僅使用蛋白質序列作為起始量。具體來說,它通過掃描序列生成結構域信息,通過預先訓練的蛋白質語言模型提取殘基特征,并根據預測的結構構建結構圖。
由于蛋白質在細胞環境中執行功能,因此它們的功能會隨著環境而動態改變。如何準確預測動態函數是未來需要解決的另一個挑戰。
原文鏈接:https://www.nature.com/articles/s41467-024-54816-8
源代碼:https://github.com/CSUBioGroup/DPFunc
相關推薦




- 免責聲明
- 本文所包含的觀點僅代表作者個人看法,不代表新火種的觀點。在新火種上獲取的所有信息均不應被視為投資建議。新火種對本文可能提及或鏈接的任何項目不表示認可。 交易和投資涉及高風險,讀者在采取與本文內容相關的任何行動之前,請務必進行充分的盡職調查。最終的決策應該基于您自己的獨立判斷。新火種不對因依賴本文觀點而產生的任何金錢損失負任何責任。
