

 新火種
2024-11-28
新火種
2024-11-28 


壓縮率達(dá)10的48次方,實(shí)現(xiàn)蛋白序列空間極端壓縮,清華EvoAI登Nature子刊

編輯 | 蘿卜皮
設(shè)計(jì)功能更佳的蛋白質(zhì)需要深入了解序列和功能之間的關(guān)系,這是一個(gè)難以探索的廣闊空間。通過識(shí)別功能上重要的特征來有效壓縮這一空間的能力極其寶貴。
清華大學(xué)的研究團(tuán)隊(duì)建立了一種稱為 EvoScan 的方法,用于全面分割和掃描高適應(yīng)度序列空間,以獲得能夠捕捉其基本特征(尤其是在高維度中)的錨點(diǎn)。
該方法適用于任何能夠與轉(zhuǎn)錄輸出耦合的生物分子功能研究。
然后,研究人員開發(fā)深度學(xué)習(xí)和大型語言模型,以從這些錨點(diǎn)準(zhǔn)確地重建空間,從而無需先前的同源性或結(jié)構(gòu)信息即可計(jì)算預(yù)測(cè)新的、高度擬合的序列。
他們將這種混合實(shí)驗(yàn)計(jì)算方法(稱為 EvoAI)應(yīng)用于阻遏蛋白,發(fā)現(xiàn)僅 82 個(gè)錨點(diǎn)就足以壓縮高適應(yīng)度序列空間,壓縮率為 1048。
該研究以「EvoAI enables extreme compression and reconstruction of the protein sequence space」為題,于 2024 年 11 月 11 日發(fā)布在《Nature Methods》。

蛋白質(zhì)工程和設(shè)計(jì)可以創(chuàng)建具有優(yōu)化功能的蛋白質(zhì),用于生物技術(shù)、醫(yī)學(xué)和合成生物學(xué)的各種應(yīng)用。
蛋白質(zhì)工程的基本挑戰(zhàn)是理解和操縱蛋白質(zhì)適應(yīng)度景觀,這是一個(gè)高維且復(fù)雜的空間,包含大量可能的序列和功能。
盡管過去幾十年來科學(xué)家們進(jìn)行了大量嘗試來尋找這個(gè)空間中的高適應(yīng)度序列,但我們對(duì)這個(gè)空間的規(guī)則和特征的理解仍然比較淺層。
雖然現(xiàn)有的定向進(jìn)化技術(shù)能提供有價(jià)值的信息,但這些實(shí)驗(yàn)方法需要在精確度與覆蓋度之間進(jìn)行權(quán)衡,且通常無法充分考慮高維空間中的自然選擇壓力。
計(jì)算方法,如基于結(jié)構(gòu)或序列的建模,雖然能評(píng)估更大的序列空間,但受限于訓(xùn)練數(shù)據(jù)的可用性,且往往忽略了生物學(xué)因素。
EvoAI
理想的方案應(yīng)結(jié)合高通量實(shí)驗(yàn)數(shù)據(jù)和高效的計(jì)算模型,通過識(shí)別「錨點(diǎn)」來壓縮設(shè)計(jì)空間,為深度學(xué)習(xí)模型提供指導(dǎo),探索整個(gè)適應(yīng)性景觀。
在最新的研究中,清華大學(xué)的研究人員開發(fā)了 EvoAI,這是一種經(jīng)驗(yàn)性地詢問序列空間,然后對(duì)其進(jìn)行建模、壓縮和重建的方法。該方法結(jié)合了高通量實(shí)驗(yàn)進(jìn)化和計(jì)算方法來捕捉和學(xué)習(xí)空間的基本特征。
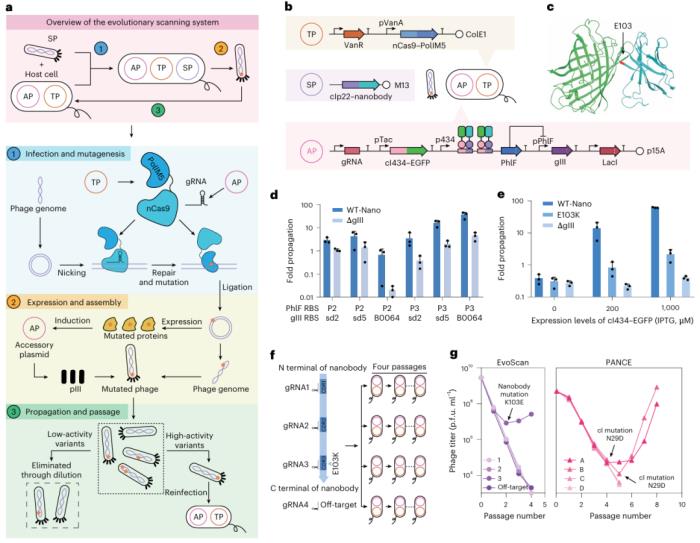
圖示:EvoScan 方案,蛋白質(zhì)-蛋白質(zhì)相互作用進(jìn)化的開發(fā)和驗(yàn)證。(來源:論文)
研究人員首先開發(fā)了一種進(jìn)化掃描方法,該方法通過結(jié)合基于EvolvR的分段誘變系統(tǒng)來適應(yīng)噬菌體輔助非連續(xù)進(jìn)化 (PANCE)。
然后,團(tuán)隊(duì)開發(fā)了一個(gè)深度學(xué)習(xí)和大型語言模型,從這些錨點(diǎn)重建序列空間并設(shè)計(jì)新的蛋白質(zhì),所有十種蛋白質(zhì)都表現(xiàn)出比野生型(WT)大大提高的活性(高達(dá) 11 倍)。
研究人員將 EvoAI 應(yīng)用于阻遏蛋白,結(jié)果顯示僅 82 個(gè)錨點(diǎn)就足以壓縮高適應(yīng)度序列空間,壓縮率為 1048。
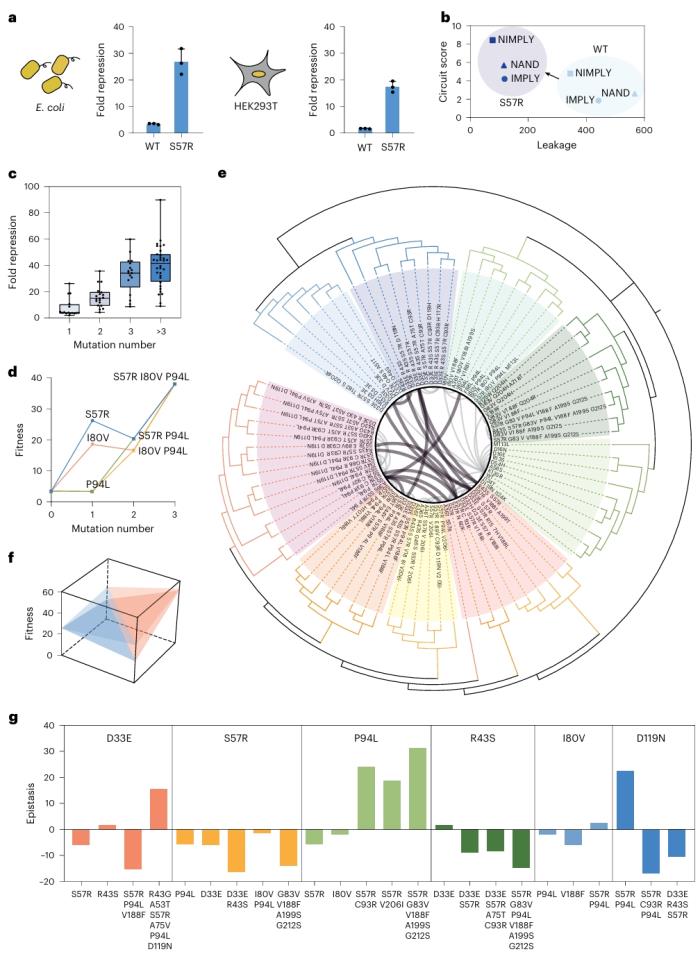
圖示:EvoScan 生成的 82 個(gè)錨點(diǎn)的遺傳關(guān)系和特征。(來源:論文)
幾個(gè)重要優(yōu)勢(shì)
與現(xiàn)有方法相比,該方法有幾個(gè)重要優(yōu)勢(shì)。
首先,它平衡了現(xiàn)實(shí)的適應(yīng)度優(yōu)化和序列空間的均勻采樣,可以快速探索高維度,生成更多樣化和功能性的變體,并提供有關(guān)序列功能關(guān)系的更豐富信息。
其次,通過在 EvoAI 中集成經(jīng)驗(yàn)進(jìn)化掃描和深度學(xué)習(xí)模型,它可以利用兩種方法的優(yōu)勢(shì)。它可以利用深度學(xué)習(xí)學(xué)到的屬性來動(dòng)態(tài)地指導(dǎo)掃描過程。可解釋的深度學(xué)習(xí)的未來發(fā)展可以揭示潛在的規(guī)則或模式,并深入了解蛋白質(zhì)如何適應(yīng)和克服進(jìn)化的限制或權(quán)衡。
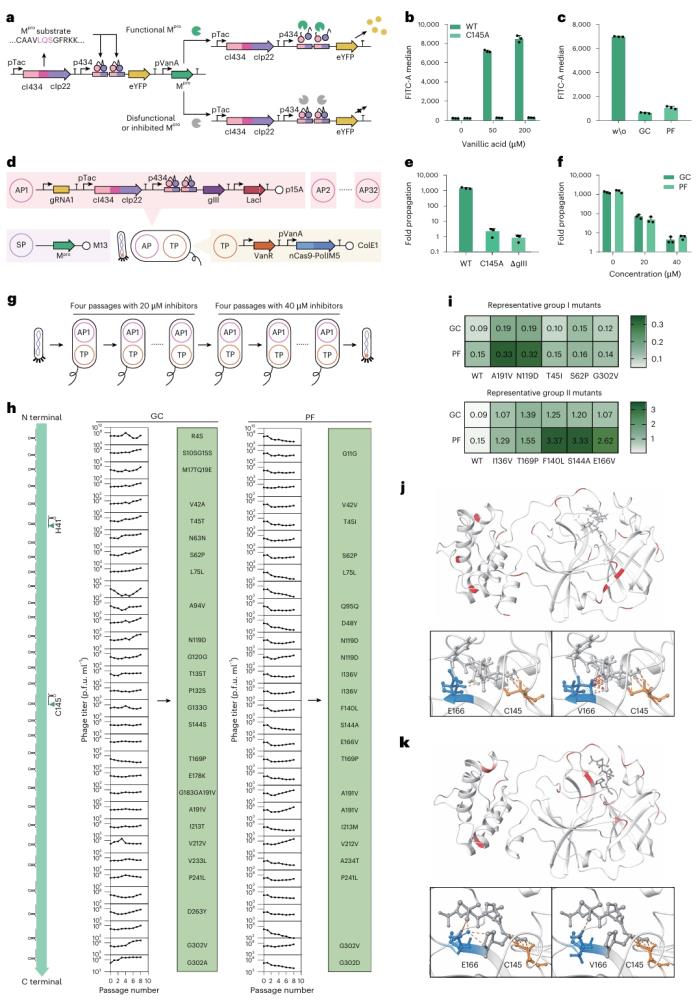
圖示:徹底的片段掃描以了解蛋白質(zhì)-配體相互作用的演變。(來源:論文)
第三,該方法可以拓展和研究缺乏結(jié)構(gòu)信息或涉及具有挑戰(zhàn)性的相互作用的蛋白質(zhì)。實(shí)驗(yàn)表明,EvoScan 可以捕獲具有多種功能的蛋白質(zhì)的錨點(diǎn),例如蛋白質(zhì)-蛋白質(zhì)、蛋白質(zhì)-配體和蛋白質(zhì)-核酸相互作用。
理論上,該方法應(yīng)該與任何可以與轉(zhuǎn)錄輸出耦合的生物分子功能兼容(例如,通過小分子傳感器的酶),因此可以應(yīng)用于研究各種生物分子的序列空間。
同時(shí),研究人員表示,研究中測(cè)試的化學(xué)空間(例如酶反應(yīng))或配體空間(例如蛋白質(zhì)相互作用)可能有所不同,需要進(jìn)一步探索。
優(yōu)化方向與展望
研究人員表示,該方法未來可以進(jìn)一步優(yōu)化改進(jìn)。
接下來,研究人員可以使用具有更多原型間隔區(qū)相鄰基序選項(xiàng)的 Cas9 變體來增加gRNA平鋪和突變靶向片段的選擇。它們還可以修改編輯系統(tǒng),一次性在多個(gè)位點(diǎn)引入突變,避免宿主切換并加快探索過程。
此外,將 EvoScan 的靶向誘變方法整合到 PACE 中可能會(huì)實(shí)現(xiàn)對(duì)序列空間片段的更深入采樣。并且,將 EvoScan 與 Evoracle 等基因型重建方法相結(jié)合,可以更系統(tǒng)、更智能地探索序列空間。
另外,該系統(tǒng)的模塊化使其非常適合自動(dòng)化,例如最近報(bào)道的 PRANCE 方法,并且可以擴(kuò)大規(guī)模以提供不同蛋白質(zhì)靶標(biāo)更全面的適應(yīng)度景觀分析數(shù)據(jù),說明高適應(yīng)度基因型的設(shè)計(jì)空間的極端壓縮性是普遍的還是不尋常的,或者整個(gè)蛋白質(zhì)適應(yīng)度景觀是否可壓縮。
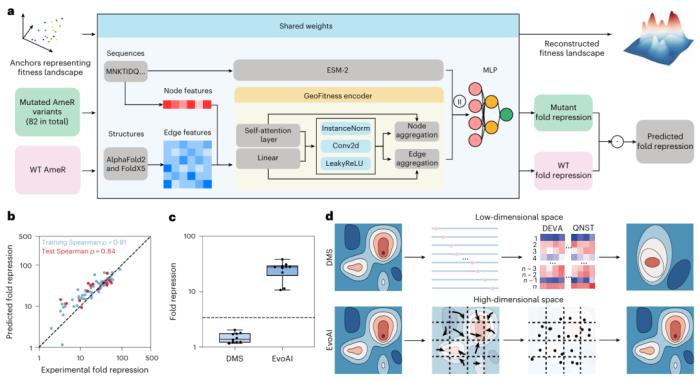
研究人員還希望他們的方法能夠激發(fā)人們對(duì)基因型和表型之間的關(guān)系以及生物系統(tǒng)進(jìn)化的新見解。設(shè)計(jì)空間的可壓縮性可能表明,大自然以某種方式找到了一種方法,通過達(dá)爾文進(jìn)化論在地球上相對(duì)較短的生命期內(nèi)搜索看似無限的空間。大型有性群體中染色體區(qū)域的基因重組可能使這種維度壓縮從而促進(jìn)進(jìn)化。
然而,重要的是要認(rèn)識(shí)到自然進(jìn)化是多方面的,受到不同的選擇壓力、波動(dòng)的環(huán)境條件、條件中性和基因組背景的影響——這些因素在目前的方法中尚未完全考慮。
該方法將來與高通量實(shí)驗(yàn)相結(jié)合,可能有助于研究生物系統(tǒng)進(jìn)化結(jié)果的路徑依賴性,并為生物技術(shù)和生物醫(yī)學(xué)應(yīng)用中的進(jìn)化和蛋白質(zhì)設(shè)計(jì)提供寶貴的見解。
論文鏈接:https://www.nature.com/articles/s41592-024-02504-2
Tags:
相關(guān)推薦




- 免責(zé)聲明
- 本文所包含的觀點(diǎn)僅代表作者個(gè)人看法,不代表新火種的觀點(diǎn)。在新火種上獲取的所有信息均不應(yīng)被視為投資建議。新火種對(duì)本文可能提及或鏈接的任何項(xiàng)目不表示認(rèn)可。 交易和投資涉及高風(fēng)險(xiǎn),讀者在采取與本文內(nèi)容相關(guān)的任何行動(dòng)之前,請(qǐng)務(wù)必進(jìn)行充分的盡職調(diào)查。最終的決策應(yīng)該基于您自己的獨(dú)立判斷。新火種不對(duì)因依賴本文觀點(diǎn)而產(chǎn)生的任何金錢損失負(fù)任何責(zé)任。
