

 新火種
2024-11-15
新火種
2024-11-15 


不懂AI、不會編碼?如何輕松拿捏AlphaFold準確預測蛋白結構

編輯 | 蘿卜皮
自 2021 年公開發布以來,用 AlphaFold2 (AF2) 預測蛋白質結構研究生物學問題,已經成為一種常見做法。
ColabFold-AF2 是 Google Colaboratory 內部的開源Jupyter Notebook,也是一個命令行工具,可讓你輕松使用 AF2,同時展示了其高級選項。
ColabFold-AF2 優化了 AF2 模型的使用,縮短了實驗的周轉時間。
在這里,韓國首爾國立大學的研究團隊發布了一項 protocol,可以幫助研究人員更方便簡潔的使用 AF2,同時介紹了一些操作技巧。
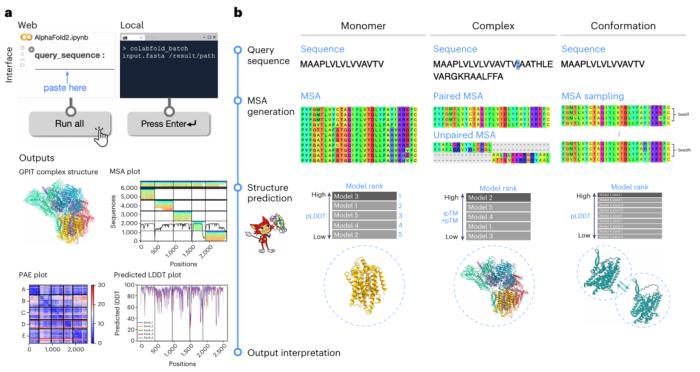
在此protocol中,研究人員通過以下三種場景引導讀者了解 ColabFold 的最佳實踐:(i)單體預測,(ii)復合物預測和(iii)構象采樣。
前兩種情況涵蓋了經典的靜態結構預測,并在人類糖基磷脂酰肌醇轉酰胺酶蛋白上進行了演示。第三種場景通過預測人類丙氨酸絲氨酸轉運蛋白 2 的兩種構象展示了 AF2 模型的另一種用例。
用戶可以通過 Google Colaboratory 運行該 protocol,而無需計算專業知識,高級用戶也可以在命令行環境中運行該 protocol。
該研究以「Easy and accurate protein structure prediction using ColabFold」為題,于 2024 年 10 月 14 日發布在《Nature Protocols》。

關于 AlphaFold
僅從蛋白質序列預測其三維結構長期以來一直是結構生物學領域的一項艱巨任務。機器學習模型的進步在實現這一目標方面取得了重大進展。
AlphaFold2 (AF2) 和RoseTTAFold代表了這些突破性的模型。它們首次提供了計算方法,能夠在提供足夠的序列信息的情況下生成與實驗解決的結構幾乎無法區分的蛋白質結構預測。具體來說,AF2 是一個端到端神經網絡,由兩個主要模塊組成。
第一個模塊處理有關輸入氨基酸 (AA) 序列 (查詢) 的信息,并根據多序列比對 (MSA) 生成關于哪些 AA 相互接觸的假設。第二個模塊匯總這些假設以預測結構(即每個原子的 3D 坐標)。
AF2 網絡背后的兩個關鍵思想是使用深度學習注意機制,這使網絡能夠更好地識別接觸的 AA,并通過多次通過模塊來細化預測。類似的原則指導了 RoseTTAFold 的設計,從而產生了一種不同的網絡架構,其卓越準確性可與 AF2 相媲美。
AF2 最初設計用于預測單鏈(單個蛋白質鏈)的結構。然而,其模型已成功用于預測多鏈或復合物之間的相互作用。
此外,AF2 模型得到了進一步開發和訓練,專門針對多聚體輸入,從而產生了 AlphaFold-multimer。從那時起,已經開發了許多其他使用 AF2 或其概念作為基礎的模型。
關于 ColabFold
ColabFold 是一個集成的蛋白質預測解決方案,旨在簡化用戶的結構建模過程。因此,它既提供了各種蛋白質預測模型的簡單界面,也提供了預處理和后處理程序。ColabFold 有兩個界面:基于 Web 的界面(使用 Google Colaboratory notebooks,以下簡稱 Colab)和命令行工具。
其基于 Web 的界面包括五個 notebooks:AlphaFold2.ipynb (用于使用 AF2 或 AF2-multimer)、RoseTTAFold2.ipynb、RoseTTAFold.ipynb(主要用于舊版)、ESMFold.ipynb 和 OmegaFold.ipynb(在下面的 ColabFold-AF2 替代品中簡要介紹)。基于 Web 的界面需要免費注冊 Colab,主要用于進行單個或小批量預測。
命令行界面僅包含 AF2 和 AF2-multimer 預測模型,并允許通過處理多個輸入序列進行批量預測。由于 AF2 模型是目前發布的最準確的模型之一,因此 protocol 專注于 AlphaFold2.ipynb 和命令行界面,它們統稱為「ColabFold-AF2」。
由于其簡單性和功能性,ColabFold 已被廣泛用于眾多研究,其公共 MSA 服務器每天被使用數萬次。它的適用性涵蓋了許多生物學領域。該論文將指導讀者如何使用 ColabFold-AF2 解決類似的生物學問題。
新的 protocol
在 protocol 中,首爾國立大學的研究人員修改并擴展了 del Alamo 等人提出的 protocol,并通過使用人類丙氨酸絲氨酸轉運蛋白 2(ASCT2,一種 Na+ 獨立的中性 AA 轉運蛋白)來展示這種能力。
該轉運蛋白是一種同型三聚體,至少有兩種構象,具體取決于它是面向細胞外(向外)還是細胞內(向內)。
通過調整 ColabFold-AF2,該團隊展示了預測這些不同結構狀態的能力。
論文概述了使用 ColabFold-AF2 進行單體(程序 1、2)和復合物(程序 3、4)預測以及構象采樣(程序 5、6)的臨時方法的綜合方案。
程序 1、3、5 是使用 Colab 的基于 Web 的方法,程序 2、4 、6 是使用命令行界面的本地方法。

ColabFold-AF2 允許通過單擊進行蛋白質預測,稱為「快速啟動」。要快速啟動,請打開 Web 瀏覽器并導航到 https://alphafold.colabfold.com。接下來,將目標蛋白質的 AA 序列粘貼到 query_sequence 字段中,然后單擊菜單欄中的 Runtime → Run all(下圖)。
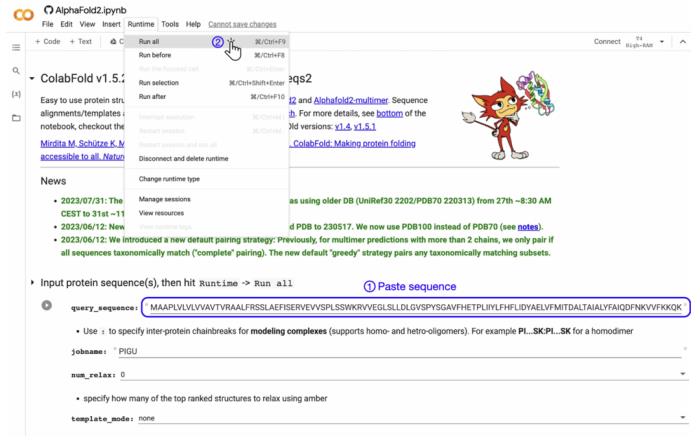
值得注意的是,如果連續運行多個預測,請單擊「重新啟動會話并運行全部」而不是「運行全部」,以避免任何沖突或內存泄漏。這將連續執行 Notebook 中的每個單元而不間斷,并且當前正在運行的單元由左側的旋轉圓圈表示。默認情況下,ColabFold 會計算五個結構模型。
生成每個模型后,會顯示預測的結構和結果圖。處理完所有單元格后,將出現一個彈出窗口,提示你下載各種結果文件作為單個壓縮 (zip) 文件。請注意,這些步驟的運行時間可能因 Colab 分配的 GPU 而異。
使用 protocol 的命令行版本需要對 Unix/Linux shell有基本的了解,并且需要能夠處理 AF2 模型的工作站。
研究人員使用人類 GPIT 蛋白演示程序 1-4。程序 1 和 2 將其五個亞基 PIGU、PIGK、PIGT、PIGS 和 GPAA1 中的每一個預測為單體,程序 3 和 4 將它們聯合預測為復合物。在程序 5 和 6 中,他們使用人類 ASCT2。
為了區分方便,研究人員根據參數的作用將其分為幾類。但是,這些參數分布在 Notebook 的多個單元格中。在每個過程中,論文詳細介紹了用戶如何向 ColabFold 提供輸入蛋白質序列,BOX3 提供了有關 ColabFold 可接受的輸入和輸出格式的完整詳細信息。

BOX4 提供了 ColabFold-AF2 計算的各種置信度測量的信息,BOX5 適用于不想使用默認 ColabFold 服務器進行 MSA 計算的用戶。


此 protocol 的預期結果部分包含有關解釋 ColabFold 圖表和輸出的一般說明,然后演示了每個程序示例的解釋過程。此 protocol 主要面向旨在進行結構分析的生物學家,不需要編碼專業知識。
詳細信息請閱讀原論文。
論文鏈接:https://www.nature.com/articles/s41596-024-01060-5
相關推薦


- 免責聲明
- 本文所包含的觀點僅代表作者個人看法,不代表新火種的觀點。在新火種上獲取的所有信息均不應被視為投資建議。新火種對本文可能提及或鏈接的任何項目不表示認可。 交易和投資涉及高風險,讀者在采取與本文內容相關的任何行動之前,請務必進行充分的盡職調查。最終的決策應該基于您自己的獨立判斷。新火種不對因依賴本文觀點而產生的任何金錢損失負任何責任。
