

 新火種
2025-04-09
新火種
2025-04-09 


Nature子刊,字節跳動開發MD模擬預測框架,助力鋰電池液體電解質研究

編輯 | 蘿卜皮
盡管機器學習力場 (MLFF) 在固體和小分子中得到廣泛應用,但在應用 MLFF 模擬液體電解質方面仍存在明顯差距——液體電解質是當前商用鋰離子電池的關鍵組成部分。
在這里,字節跳動團隊的研究人員提出了 ByteDance AI Molecular Simulation Booster (BAMBOO),這是一種用于分子動力學(MD)模擬的預測框架,并展示了其在鋰電池液體電解質環境中的預測能力。
研究人員設計了一個受物理啟發的圖等變 Transformer 架構(GET)作為 BAMBOO 的主干,以便從量子力學模擬中學習;同時,他們引入了一種集成知識蒸餾方法并將其應用于 MLFF,以減少分子動力學模擬觀測值的波動。
他們還提出了一種密度對齊算法,從而使 BAMBOO 與實驗測量值對齊。BAMBOO 在預測各種溶劑和鹽組合中的關鍵電解質特性(例如密度、粘度和離子電導率)方面表現出了最先進的準確性。
當前版本的模型針對 15 種化學物質進行了訓練,與實驗相比,各種成分的平均密度誤差為 0.01?g·cm^-3。
該研究以「A predictive machine learning force-field framework for liquid electrolyte development」為題,于 2025 年 4 月 1 日發布在《Nature Machine Intelligence》。

液態電解質是目前大多數商用鋰離子電池中不可或缺的組成部分。現有的商用電解質多為碳酸鹽基,通常由五種以上的成分組成以滿足不同的需求。通過實驗探索分子相互作用以合理設計電解質成本高昂、耗時長,并且嚴重依賴化學家的直覺和經驗。
這些限制對從實驗室的概念驗證實驗過渡到市場產品帶來了挑戰,特別是由于優化多組分液態電解質性能的復雜性呈指數級增長。
當前,比較有潛力的解決方案是用機器學習力場(MLFFs)對液態電解質進行模擬,不過該方法也面臨著雙重挑戰:
一方面需平衡量子力學精度與經典力場效率,通過圖神經網絡與物理驅動模型融合提升通用性,但離子-溶劑復雜結構(如 SSIP/CIP/AGG 共存)導致跨體系預測困難;
另一方面,現有研究雖驗證了特定體系可行性,卻缺乏普適性證據,且 MLFFs 普遍存在模擬崩潰、結果波動及過度依賴量子數據而偏離實驗的問題,實驗數據驅動的參數優化因計算成本與過擬合風險尚未有效實現。
BAMBOO
在最新的工作中,字節跳動的研究人員介紹了 BAMBOO(ByteDance AI Molecular Simulation Booster)工作流程,該工作流程專門用于構建用于有機液體 MD 模擬的 MLFF,特別是液體電解質。
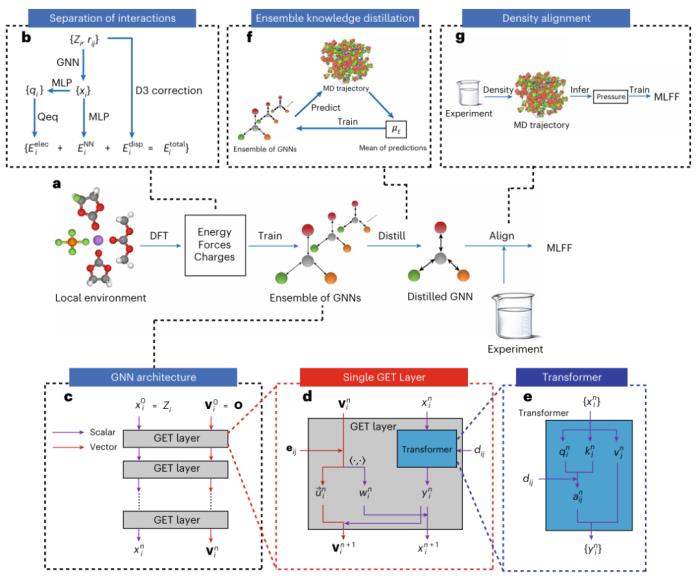
圖示: BAMBOO 概述。(來源:論文)
首先,研究人員設計了一個 GET 架構,利用 DFT 計算的知識,將半局部、靜電和色散相互作用分開;具體來說,他們將液體電解質中的局部原子環境采樣為氣相團簇,然后使用 DFT 計算它們的能量、原子力和電荷。
其次,他們在 MLFF 上引入了集成知識蒸餾,抑制了基于 MLFF 的 MD 模擬結果的波動。同時研究人員提出了一種基于物理的對齊方法來協調模擬密度與實驗數據,從而建立宏觀和微觀尺度之間的聯系。
圖示:GET 層的影響、集成知識提煉和密度對齊。(來源:論文)
為了保證 BAMBOO 的廣泛適用性,該團隊在 DFT 數據集中包含了各種分子和鹽。值得注意的是,他們的重點涵蓋了鋰離子電池中使用的液體電解質中常見的成分,例如環狀碳酸酯、線性碳酸酯以及 Li+ 陽離子和 PF6-、FSI- 和 TFSI- 陰離子。
此外,研究人員結合了兩種常用的有機液體乙醇 (EO) 和丙酮 (ACT),以及工程流體 Novec7000,從而確保訓練模型的通用性。
評估結果
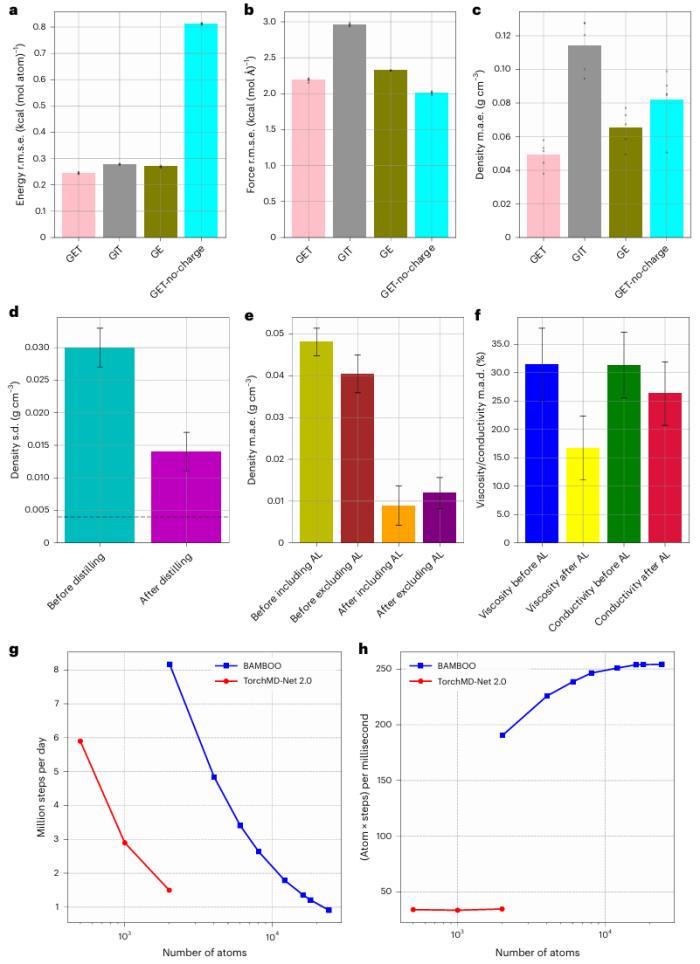
研究人員對 BAMBOO 在溶劑和液體電解質上的性能進行了全面評估。模擬結果表明,單個 BAMBOO 模型可以高精度地預測各種化學物質的密度、粘度和離子電導率。
當前的 BAMBOO 模型能夠模擬多達 15 種不同成分的物種。這種穩健性凸顯了 BAMBOO 在促進實際液體電解質設計和優化方面的價值,這些電解質可能包含多達 10 種成分。
除了預測本體特性的最先進的準確性之外,研究人員還展示了溶劑化結構和原子部分電荷與電解質成分之間關系的量化,為溶劑化工程提供了經典力場或 DFT 無法得出的解析。
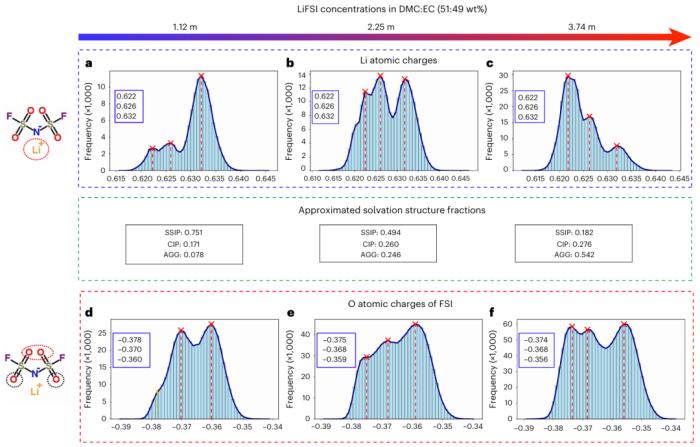
圖示:使用 BAMBOO 模擬三種 LiFSI 電解質的原子電荷分布和溶劑化結構分數。(來源:論文)
總而言之,BAMBOO 在預測各種液體電解質的密度、粘度和離子電導率方面達到了最先進的精度。它的高預測能力使其成為由分子結構工程驅動的電解質設計的寶貴工具。
與目前的經典(可極化)力場相比,由于 BAMBOO 是一種沒有明確限制函數形式的機器學習力場,因此它有可能擴展到模擬體積特性以外的其他領域,例如模擬液體中的化學反應。
研究人員設想這項工作為開發能夠準確模擬大多數有機液體的特性和行為的「通用機器學習力場」奠定了基礎。
論文鏈接:https://www.nature.com/articles/s42256-025-01009-7
相關推薦


- 免責聲明
- 本文所包含的觀點僅代表作者個人看法,不代表新火種的觀點。在新火種上獲取的所有信息均不應被視為投資建議。新火種對本文可能提及或鏈接的任何項目不表示認可。 交易和投資涉及高風險,讀者在采取與本文內容相關的任何行動之前,請務必進行充分的盡職調查。最終的決策應該基于您自己的獨立判斷。新火種不對因依賴本文觀點而產生的任何金錢損失負任何責任。
