

 新火種
2023-12-21
新火種
2023-12-21 


人工智能為新藥鋪平道路:幾何深度學(xué)習(xí)方法可以預(yù)測(cè)合成藥物分子的最佳方案

編輯 | 蘿卜皮
后期功能化是優(yōu)化候選藥物特性的一種經(jīng)濟(jì)方法。然而,藥物分子的化學(xué)復(fù)雜性往往使得后期多樣化具有挑戰(zhàn)性。
為了解決這個(gè)問(wèn)題,德國(guó)慕尼黑大學(xué)(Ludwig-Maximilians-Universit?t München)、蘇黎世聯(lián)邦理工學(xué)院(ETH Zurich)和巴塞爾羅氏制藥(Roche Innovation Center Basel)的研究人員開(kāi)發(fā)了基于幾何深度學(xué)習(xí)和高通量反應(yīng)篩選的后期功能化平臺(tái)。
考慮到硼基化是后期功能化的關(guān)鍵步驟,計(jì)算模型預(yù)測(cè)了不同反應(yīng)條件下的反應(yīng)產(chǎn)率,平均絕對(duì)誤差范圍為 4-5%;模型對(duì)已知和未知底物的新反應(yīng)的反應(yīng)性進(jìn)行分類(lèi),平衡準(zhǔn)確度分別為 92% 和 67%。主要產(chǎn)物的區(qū)域選擇性被準(zhǔn)確捕獲,分類(lèi)器 F 分?jǐn)?shù)為 67%。當(dāng)應(yīng)用于 23 種不同的商業(yè)藥物分子時(shí),該平臺(tái)成功發(fā)現(xiàn)了許多結(jié)構(gòu)多樣化的機(jī)會(huì)。
該研究以「Enabling late-stage drug diversification by high-throughput experimentation with geometric deep learning」為題,于 2023 年 11 月 23 日發(fā)布在《Nature Chemistry》。

LSF項(xiàng)目在藥物化學(xué)研究中有著舉足輕重的作用
當(dāng)旨在建立藥物化學(xué)中的結(jié)構(gòu)-活性關(guān)系時(shí),結(jié)構(gòu)的新穎性和復(fù)雜性使得化學(xué)目標(biāo)結(jié)構(gòu)的合成具有挑戰(zhàn)性。構(gòu)效關(guān)系模型指導(dǎo)先導(dǎo)化合物和先導(dǎo)化合物優(yōu)化方案,旨在提高候選藥物的藥理活性和理化性質(zhì)。對(duì)于結(jié)構(gòu)-活性關(guān)系探索,高效的整合非常重要,這是設(shè)計(jì)-制造-測(cè)試-分析周期的瓶頸。
許多選擇性激活和修飾 C-H 鍵的合成方法可實(shí)現(xiàn)有機(jī)支架的后期功能化 (LSF),范圍從分子構(gòu)件到高級(jí)藥物分子。許多催化系統(tǒng)提供定向和非定向方法,以及對(duì)修飾類(lèi)似物的化學(xué)和位點(diǎn)選擇性訪(fǎng)問(wèn)。
在眾多 LSF 方法中,C-H 硼化被認(rèn)為是最通用的快速化合物多樣化方法。有機(jī)硼物質(zhì)可以轉(zhuǎn)化為一系列官能團(tuán),并作為后續(xù) C-C 鍵偶聯(lián)的穩(wěn)健手柄,從而實(shí)現(xiàn)廣泛的結(jié)構(gòu)-活性關(guān)系研究。
然而,迄今為止,LSF 在藥物發(fā)現(xiàn)中的應(yīng)用僅有少數(shù)報(bào)道。大多數(shù)這些罕見(jiàn)的例子都集中在單一的 LSF 反應(yīng)類(lèi)型上。多個(gè)官能團(tuán)和具有不同鍵強(qiáng)度、電子特性以及空間和官能團(tuán)環(huán)境的各種類(lèi)型的 C-H 鍵對(duì)直接 LSF 提出了挑戰(zhàn)。并且開(kāi)展 LSF 項(xiàng)目通常非常耗時(shí)且需要資源密集的實(shí)驗(yàn),這與許多藥物化學(xué)項(xiàng)目的緊迫時(shí)間表和有限資產(chǎn)不相容。
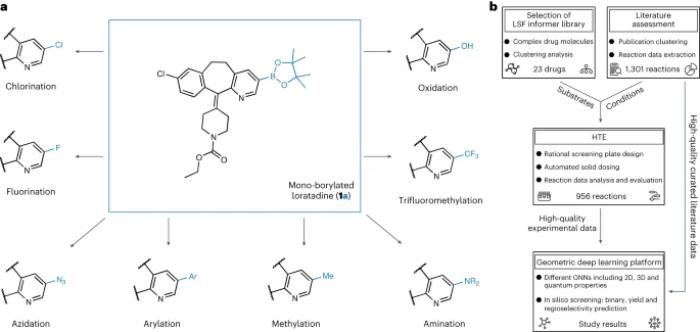
圖示:硼化多樣化研究概述。(來(lái)源:論文)
人工智能支持的 LSF
高通量實(shí)驗(yàn) (HTE) 是一種既定的反應(yīng)優(yōu)化方法,可實(shí)現(xiàn)半自動(dòng)小型化小批量篩選,從而快速、可重復(fù)地使用少量珍貴的構(gòu)建模塊和耗材并行執(zhí)行多個(gè)轉(zhuǎn)化。結(jié)合可生成有關(guān)成功和失敗反應(yīng)的高質(zhì)量數(shù)據(jù)集的 FAIR(可查找性、可訪(fǎng)問(wèn)性、互操作性、可重用性)文檔,HTE 通過(guò)實(shí)現(xiàn)高級(jí)數(shù)據(jù)分析和機(jī)器學(xué)習(xí),為解鎖 LSF 進(jìn)行藥物發(fā)現(xiàn)奠定了基礎(chǔ)。
圖神經(jīng)網(wǎng)絡(luò)(GNN)在分子特征提取和屬性預(yù)測(cè)方面有著廣泛的應(yīng)用。在為化學(xué)反應(yīng)規(guī)劃開(kāi)發(fā)的各種機(jī)器學(xué)習(xí)方法中,GNN 已成功應(yīng)用于逆合成規(guī)劃、區(qū)域選擇性預(yù)測(cè)和反應(yīng)產(chǎn)物預(yù)測(cè)。此外,還開(kāi)發(fā)了 transformer 和基于指紋的方法來(lái)解決類(lèi)似的問(wèn)題。
有研究表明,學(xué)習(xí)過(guò)渡態(tài)幾何結(jié)構(gòu)的活化能可以準(zhǔn)確預(yù)測(cè)競(jìng)爭(zhēng)反應(yīng)結(jié)果。使用密度泛函理論(DFT)級(jí)原子部分電荷的圖形特征化改進(jìn)了對(duì)電子效應(yīng)驅(qū)動(dòng)的反應(yīng)的區(qū)域選擇性的預(yù)測(cè)。圖機(jī)器學(xué)習(xí)與 HTE 的結(jié)合可以?xún)?yōu)化有機(jī)底物 C-H 活化的反應(yīng)條件。有些研究重點(diǎn)關(guān)注使用過(guò)渡態(tài)的深度學(xué)習(xí)模型,這類(lèi)模型具有預(yù)測(cè)反應(yīng)結(jié)果的能力,包括在某些情況下的對(duì)映選擇性。
然而,這些方法僅限于小分子結(jié)構(gòu)和相對(duì)較小的數(shù)據(jù)集,使得將此類(lèi)模型應(yīng)用于結(jié)構(gòu)更復(fù)雜的藥物樣分子具有挑戰(zhàn)性。文獻(xiàn)里的研究表明,通過(guò)過(guò)渡態(tài)的量子化學(xué)信息增強(qiáng)的混合機(jī)器學(xué)習(xí)模型,能夠?qū)︺灤呋呐鸹磻?yīng)進(jìn)行區(qū)域選擇性預(yù)測(cè)。但是,空間效應(yīng)和電子效應(yīng)對(duì) C-H 活化反應(yīng)模型性能的影響,及其在具有多個(gè)芳環(huán)系統(tǒng)的分子區(qū)域選擇性中的應(yīng)用,仍未得到探索。
自動(dòng) LSF 硼化篩選的幾何深度學(xué)習(xí)
慕尼黑大學(xué)、蘇黎世聯(lián)邦理工學(xué)院和巴塞爾羅氏制藥的研究人員介紹了一種應(yīng)用于自動(dòng) LSF 硼化篩選的幾何深度學(xué)習(xí)方法,用于識(shí)別后期命中和先導(dǎo)多樣化機(jī)會(huì)。采用計(jì)算深度學(xué)習(xí)來(lái)預(yù)測(cè)復(fù)雜藥物分子 LSF 的反應(yīng)結(jié)果、產(chǎn)量和區(qū)域選擇性。
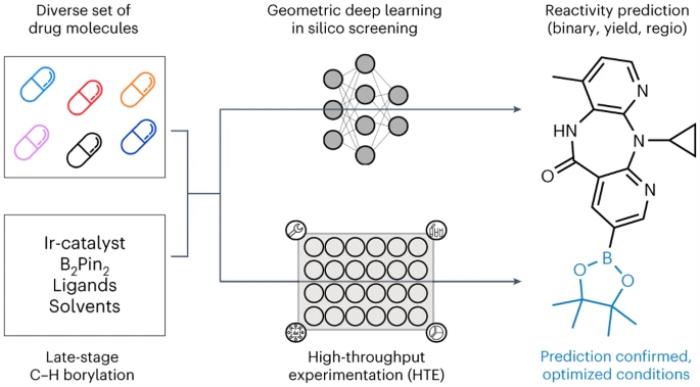
「這種方法有可能顯著減少所需的實(shí)驗(yàn)室實(shí)驗(yàn)數(shù)量,從而提高化學(xué)合成的效率和可持續(xù)性。」論文的主要作者,慕尼黑大學(xué)化學(xué)與藥學(xué)學(xué)院和羅氏公司 David Konrad 博士研究小組的博士生 David Nippa 說(shuō)。
在該研究的第一步中,對(duì)已發(fā)表的文獻(xiàn)進(jìn)行了全面分析,為選擇合適的 HTE 篩選反應(yīng)條件和反映藥物發(fā)現(xiàn)后期先導(dǎo)化合物性質(zhì)的相關(guān)底物提供了理論依據(jù)。反應(yīng)條件是根據(jù) 38 篇選定文獻(xiàn)數(shù)據(jù)集手動(dòng)整理的文獻(xiàn)數(shù)據(jù)選擇的。
LSF 底物的選擇基于對(duì) 1,174 種已批準(zhǔn)藥物的聚類(lèi)分析,產(chǎn)生了 23 種結(jié)構(gòu)不同的藥物分子。這種方法使研究人員能夠在「信息庫(kù)」方法(即涉及為評(píng)估合成方法而定制的化學(xué)空間的方法)中使用反應(yīng)條件和底物的相關(guān)示例,而不是使用適用性有限的理想化底物和片段來(lái)優(yōu)化先導(dǎo)化合物。
在第二步中,研究人員使用半自動(dòng)化 HTE 生成數(shù)據(jù)(實(shí)驗(yàn)數(shù)據(jù)集)。所選藥物分子和反應(yīng)條件的反應(yīng)數(shù)據(jù)為后續(xù)反應(yīng)結(jié)果的機(jī)器學(xué)習(xí)提供了高質(zhì)量的數(shù)據(jù)。
最后,不同的 GNN 在二維、三維和原子部分電荷增強(qiáng)分子圖上進(jìn)行訓(xùn)練,從而預(yù)測(cè)二元(是/否)反應(yīng)結(jié)果、反應(yīng)產(chǎn)率和區(qū)域選擇性。「有趣的是,當(dāng)考慮起始材料的三維信息而不僅僅是其二維化學(xué)式時(shí),預(yù)測(cè)結(jié)果會(huì)得到改善。」蘇黎世聯(lián)邦理工學(xué)院的博士生 Kenneth Atz 說(shuō)。
該方法已成功用于識(shí)別現(xiàn)有活性成分中可以引入額外活性基團(tuán)的位置。這有助于研究人員更快地開(kāi)發(fā)已知藥物活性成分的新的、更有效的變體。
Tags:
相關(guān)推薦




- 免責(zé)聲明
- 本文所包含的觀(guān)點(diǎn)僅代表作者個(gè)人看法,不代表新火種的觀(guān)點(diǎn)。在新火種上獲取的所有信息均不應(yīng)被視為投資建議。新火種對(duì)本文可能提及或鏈接的任何項(xiàng)目不表示認(rèn)可。 交易和投資涉及高風(fēng)險(xiǎn),讀者在采取與本文內(nèi)容相關(guān)的任何行動(dòng)之前,請(qǐng)務(wù)必進(jìn)行充分的盡職調(diào)查。最終的決策應(yīng)該基于您自己的獨(dú)立判斷。新火種不對(duì)因依賴(lài)本文觀(guān)點(diǎn)而產(chǎn)生的任何金錢(qián)損失負(fù)任何責(zé)任。
