

 新火種
2023-12-21
新火種
2023-12-21 


AI煉金術革新化學:MIT學者使用生成式AI,六秒生成新化學反應

編輯 |凱霞
從「等價交換」的遠古煉金術開始,化學一直是一門了解和控制物質間相互作用的學科。人們經過不斷解鎖和利用新的化學反應,研發出了一系列新材料。在為人們生活提供便利的同時也提升了能量利用效率,促進可持續發展。
一個基元化學反應由反應物,過渡態(TS),生成物三者構成。過渡態是化學中至關重要的 3D 結構,被廣泛用于理解化學反應機制、估算反應能壘以及探索龐大的反應網絡。然而,由于其在反應過程中存在的時間極短(飛秒量級),實驗中幾乎不可能分離和表征過渡態。
常規情況下,人們使用量子化學的計算方法,通過反復求解薛定諤方程,以確定已知反應物和生成物之間的過渡態。然而,這種計算方法較為昂貴,并以經常失敗而「臭名遠揚」。同時,該方法受限于個人的經驗直覺和計算所需的資源,每個人所能探索的化學反應也是局限的。這種限制在研究未知的復雜反應時尤為「致命」。它會使研究者忽略一些潛在可能發生的反應,導致會反應機理的誤判,進而影響催化材料設計的思路。
針對這一問題,麻省理工學院(MIT)的一組研究人員開發出了一種基于機器學習的替代方法,能夠在幾秒鐘內發現這些結構。他們的新模型可以協助化學家探索和設計新的反應和催化劑,從而生成高附加值的有用產品,例如燃料化合物或藥物。此外,該模型還能夠模擬自然發生的化學反應,如那些推動早期地球上生命演化的關鍵反應。
「過渡態作為設計催化劑或了解自然系統如何執行某些轉化的起點,知道其具體結構十分重要。」MIT 化學工程和化學教授 Heather Kulik 說道。
相關研究工作以「Accurate transition state generation with an object-aware equivariant elementary reaction diffusion model」為題發表在國際頂級期刊《Nature Computational Science》上。
其中麻省理工學院的段辰儒博士是第一加通訊作者。此外,康奈爾大學博士生杜沅豈,麻省理工學院博士生賈皓鈞以及麻省理工學院Heather Kulik 教授為該論文的共同作者。

MIT News 也報道了該研究。

理論難點
現階段,化學家可以使用一種基于密度泛函理論的量子化學計算方法來計算過渡態。然而,這種方法需要大量的計算資源,需要數小時甚至數天才能完成一個過渡態的計算。
為了解決計算時間久的問題,一些研究人員在近期開始嘗試使用機器學習模型來發現過渡態結構。然而,迄今為止幾乎所有開發的模型都要求將兩個反應物建模為一個整體,而反應物之間相對于彼此則保持特定的幾何構型(conformation)。任何其他可能的構型都會被機器學習模型誤認為成一個新的反應。
「如果反應物分子被旋轉,那么從原理上講,在旋轉之前和之后它們仍然可以經歷相同的化學反應。就像我們在談論電解水的時候,只會說水在特定條件下轉換為氧氣和氫氣,而不會描述這些分子的相對幾何位置。但在傳統的機器學習方法中,模型將把反應物和生成物在不同幾何位置的反應視為兩個不同的反應。這使得機器學習訓練變得更加困難,準確性也會隨之下降。」段辰儒博士表示。
擴散模型作為一個生成式模型曾被應用于圖像處理中。最近,擴散模型還被應用于 3D 分子和蛋白結構生成、蛋白質-配體對接以及基于結構的藥物設計。在這些應用中,擴散模型利用 3D special Euclidean group (SE(3)) 圖神經網絡(GNNs)來保留分子的排列、平移和旋轉對稱性。然而,由反應物、過渡態和產物組成的基元反應卻遵循「對象感知」的 SE(3) 對稱性。這是因為基元反應中三個對象之間的相互作用不是通過 3D 歐幾里得空間進行的,而是在更高維的電子勢能面(potential energy surface)的因果聯系。因此,現有的基于 SE(3) GNN 的擴散模型會因為破壞對稱性而存在問題。
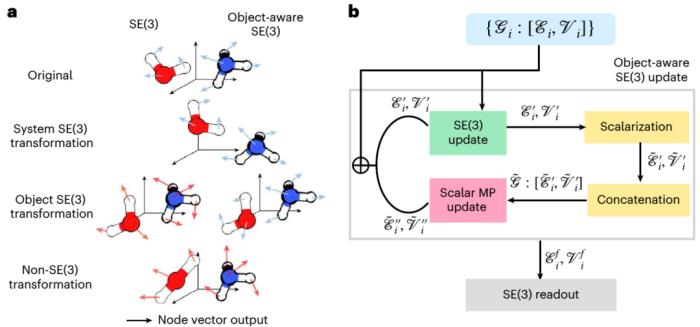
圖示:「對象感知」SE(3) 等變及其基于 SE(3) 等變 GNN 的實現。(來源:論文)
解決方案
麻省理工學院的團隊根據以上問題開發了一種新的方法,叫做「OA-ReactDiff」。該團隊將 SE(3) 等變 GNN 調整為「對象感知」模擬,即在保持單個對象的 SE(3) 等變性的同時,保持它們在歐幾里得空間中獨立的相互作用。
「擴散模型屬于生成式人工智能領域,通過隨機過程捕捉簡單分布與復雜分布之間的轉化。一旦模型學到了這三種結構如何共存的基本分布,我們可以給它新的反應物和生成物,它將嘗試生成與這些反應物和生成物相對應的過渡態結構。」段辰儒博士說。
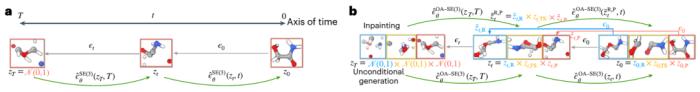
圖示:用于生成分子系統采樣的等變擴散模型 (EDM) 概述。(來源:論文)
在訓練集中,研究人員使用量子計算方法得到了 9,000 種不同化學反應的反應物、過渡態和生成物結構。并在約 1,000 個之前未見過的反應上進行了測試,要求它為每個過渡態生成 40 種可能的結構。
在計算的過程中通過引入「推薦模型」來預測哪個過渡態的置信度最高。在此基礎上進一步結合不確定性估計,研究人員在僅對 14% 模型不確定性最高的反應執行量子化學計算,就實現了 2.6 kcal/mol 的平均絕對誤差。這使得在使用 OA-ReactDiff 估算 300°C 的反應速率時,可以得到一個數量級誤差范圍內的結果。OA-ReactDiff 生成的結構與量子化學計算得到的過渡態結構相比,均方根誤差 (RMSD) 在 0.06 埃(千分之六納米)范圍內,這個誤差量級在肉眼中幾乎不可區分。
更令人欣喜的是 OA-ReactDiff 生成一個過渡態結構只需要 6 秒,相比于量子化學計算至少加速了 1000 倍。由此,該算法成功實現了對 TS 結構和反應能壘計算的極高準確性和快速性。
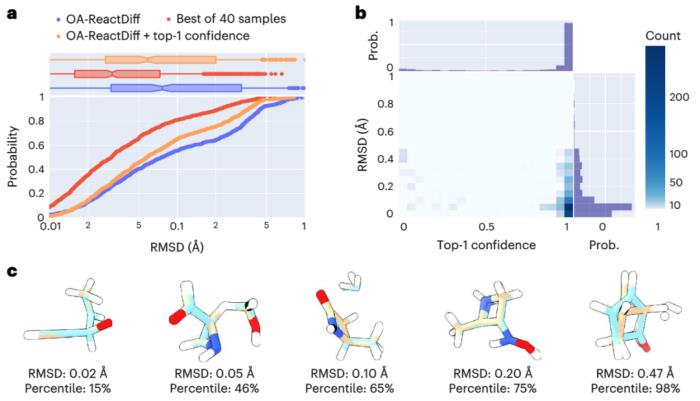
圖示:評估 OA-ReactDiff 生成的 TS 結構與真實 TS 結構的結構相似性。(來源:論文)
Kulik 教授也感慨道,「之前我們很難想象在一念之間就能生成數千個過渡態。」
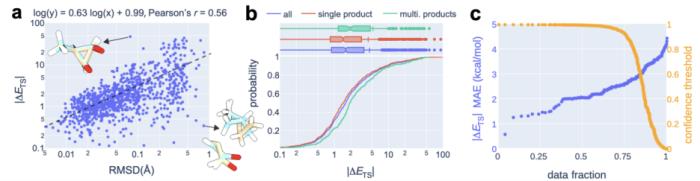
圖示:OA-ReactDiff plus 推薦 TS 結構的能量性能。(來源:論文)
未來期許
這項工作是 3D 擴散模型首次在化學反應中的亮相。盡管研究人員僅對較小數量原子的化合物(<25 個原子)的反應上進行了模擬訓練,但他們發現整個模型也能夠對較大分子的反應進行準確預測。
Kulik教授說:「即使你面對更大的系統甚至是酶催化的系統,你仍然可以得到關于原子最有可能重新排列的不同方式」。
研究人員現在計劃加入其他組分來擴展他們的模型,比如催化劑。借助生成式 AI 的隨機性,OA-ReactDiff 可以探索到意料之外的化學反應。這個特點彌補了現有基于化學的直覺反應探索框架,幫助建立更加完整的化學反應網絡,助力研發設計新型催化材料。這方面的研究可以幫助他們加速發掘特定反應的新的催化劑。此外,他們提出的算法對于開發藥品、燃料或其他有用化合物的新過程可能非常有用,尤其是當合成涉及許多化學步驟時。
「傳統上,所有這些計算都是用量子化學進行的,而現在我們能夠用更快的生成模型替代量子化學。」段辰儒博士說。
研究人員也表示化學反應是化學研究的核心。除了催化劑設計這種偏「工業型」的可能,OA-ReactDiff 還有許多有意思的潛在應用,如探索可能發生在其他行星上的氣體間相互作用,模擬地球早期生命演化過程中發生的反應過程等等。
相關推薦




- 免責聲明
- 本文所包含的觀點僅代表作者個人看法,不代表新火種的觀點。在新火種上獲取的所有信息均不應被視為投資建議。新火種對本文可能提及或鏈接的任何項目不表示認可。 交易和投資涉及高風險,讀者在采取與本文內容相關的任何行動之前,請務必進行充分的盡職調查。最終的決策應該基于您自己的獨立判斷。新火種不對因依賴本文觀點而產生的任何金錢損失負任何責任。
