

 新火種
2023-12-20
新火種
2023-12-20 


GPU上運行速度比現(xiàn)有模型快3-7倍,IU團(tuán)隊使用全卷積神經(jīng)網(wǎng)絡(luò)進(jìn)行準(zhǔn)確的從頭肽測序

編輯 | 蘿卜皮
從頭肽測序不依賴于全面的靶序列數(shù)據(jù)庫,這為科學(xué)家提供了一種從串聯(lián)質(zhì)譜中識別新肽的方法。然而,當(dāng)前的從頭測序算法的準(zhǔn)確性和覆蓋率較低,這阻礙了它們在蛋白質(zhì)組學(xué)中的應(yīng)用。
印第安納大學(xué)(Indiana University,IU)的研究人員提出了 PepNet,一種用于高精度從頭肽測序的全卷積神經(jīng)網(wǎng)絡(luò)。PepNet 將 MS/MS 譜(表示為高維向量)作為輸入,并輸出最佳肽序列及其置信度得分。PepNet 模型使用來自多個人類肽譜庫的總共 300 萬個高能碰撞解離 MS/MS 譜圖進(jìn)行訓(xùn)練。
評估結(jié)果表明,PepNet 在肽級精度和位置級精度方面均顯著優(yōu)于當(dāng)前性能最佳的從頭測序算法(例如 PointNovo 和 DeepNovo)。PepNet 可以對數(shù)據(jù)庫搜索引擎未識別的大部分光譜進(jìn)行測序,因此可以用作數(shù)據(jù)庫搜索引擎的補充工具,用于蛋白質(zhì)組學(xué)中的肽識別。
此外,PepNet 在 GPU 上的運行速度分別比 PointNovo 和 DeepNovo 快 3 倍和 7 倍,因此更適合大規(guī)模蛋白質(zhì)組數(shù)據(jù)的分析。
該研究以「Accurate de novo peptide sequencing using fully convolutional neural networks」為題,于 2023 年 12 月 2 日發(fā)布在《Nature Communications》。

過去十年見證了質(zhì)譜技術(shù)的巨大進(jìn)步,特別是液相色譜耦合串聯(lián)質(zhì)譜(LC-MS/MS)。隨著通量和靈敏度的提高,LC-MS/MS 已成為對包括人類在內(nèi)的高等生物體的各種生理(例如疾病)條件下的整個蛋白質(zhì)組規(guī)模的蛋白質(zhì)功能研究最廣泛使用的方法之一。
在典型的蛋白質(zhì)組學(xué)實驗中,獲得 MS/MS 譜圖后,第一步(可以說是最重要的步驟)是從這些譜圖中識別肽。人們開發(fā)了許多算法來解決這個問題,這些算法主要分為三類:蛋白質(zhì)數(shù)據(jù)庫搜索、光譜庫搜索和從頭測序。
蛋白質(zhì)數(shù)據(jù)庫搜索是肽鑒定的主要方法。肽序列標(biāo)簽法和 Sequest 算法是該類較早的算法。最近的開發(fā)包括 Mascot、X!Tandem、OMSSA、MyriMatch、Protein Prospector 和 MSGF+。這些方法將實驗光譜與蛋白質(zhì)數(shù)據(jù)庫中肽生成的理論光譜進(jìn)行比較,并報告那些可能真實的肽光譜匹配(PSM)。
相比之下,光譜庫搜索方法將新采集的 MS/MS 光譜與包含早期計算分析中使用的先前表征的實驗光譜的庫進(jìn)行比較。由于 MS/MS 數(shù)據(jù)的重復(fù)性和再現(xiàn)性的提高以及大量實驗光譜的可用性不斷提高(例如,來自蛋白質(zhì)組學(xué)數(shù)據(jù)存儲庫和大規(guī)模合成肽項目),光譜庫搜索方法已被越來越多地采用,并在 X!hunter、SpectraST 和 msSLASH 等軟件工具中實施。
最后,從頭測序算法嘗試直接從 MS/MS 譜圖中導(dǎo)出肽序列,而不使用譜庫或蛋白質(zhì)序列數(shù)據(jù)庫等參考資料。許多從頭測序算法采用圖論公式,通過動態(tài)規(guī)劃算法和自適應(yīng)評分方案來計算譜圖中的最長路徑。隨著高分辨率 MS 儀器的進(jìn)步,從頭測序算法的性能顯著提高,特別是在更復(fù)雜的評分方案中。
近期,DeepNovo 及其后繼模型 PointNovo 是使用深度學(xué)習(xí)算法開發(fā)的,該算法可以從大量肽的 MS/MS 譜圖中自動學(xué)習(xí)與肽序列相關(guān)的碎片離子模式,并報告了性能的改進(jìn)。這些方法利用深度神經(jīng)網(wǎng)絡(luò) (DNN) 架構(gòu)來捕獲輸入串聯(lián)質(zhì)譜中碎片離子之間的依賴性,隨后將其用于按順序構(gòu)建肽。
盡管這些方法比傳統(tǒng)的從頭測序算法表現(xiàn)出更好的性能,但研究人員觀察到它們可以對相對較少的長肽進(jìn)行測序,特別是來自 Charge 3+ MS/MS 譜圖,這可能是由于對碎片離子之間復(fù)雜的長程模式進(jìn)行建模的挑戰(zhàn)。另一方面,PredFull 采用的卷積神經(jīng)網(wǎng)絡(luò)(CNN)架構(gòu)進(jìn)行完整的 MS/MS 譜預(yù)測,展示了 CNN 學(xué)習(xí) MS/MS 譜中復(fù)雜模式的優(yōu)勢。
因此,IU 的研究人員開發(fā)了一種名為 PepNet 的深度學(xué)習(xí)模型,與之前的方法相比,該模型顯著提高了串聯(lián)質(zhì)譜從頭肽測序的性能。
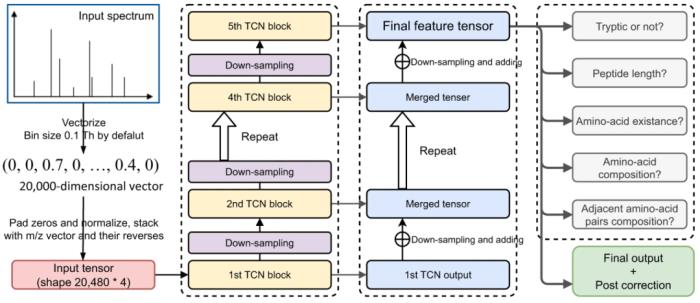
圖:PepNet 的神經(jīng)網(wǎng)絡(luò)架構(gòu)。(來源:論文)
研究人員首先證明 PepNet 能夠?qū)θ祟?MS/MS 譜圖進(jìn)行高精度測序,然后證明 PepNet 可以在來自許多非人類生物體的 MS/MS 數(shù)據(jù)中始終表現(xiàn)良好。此外,對未識別光譜的從頭測序結(jié)果表明,PepNet 有能力從 MaxQuant 忽略的光譜中發(fā)現(xiàn)大量識別,產(chǎn)生的識別比 MaxQuant 之前檢測到的識別多幾倍。
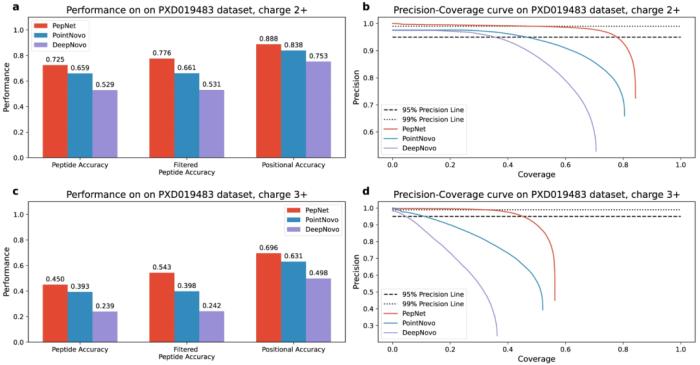
圖:PepNet、PointNovo 和 DeepNovo 在人類蛋白質(zhì)組數(shù)據(jù)集中的 2+ 和 3+ 電荷譜上的準(zhǔn)確度和精度覆蓋曲線。(來源:論文)
平均而言,在相當(dāng)?shù)木人较拢琍epNet 可以比其他工具多 2.5-19 倍地測序未識別的光譜。
這表明,盡管 PepNet 是使用 MaxQuant 等數(shù)據(jù)庫搜索工具測序的肽進(jìn)行訓(xùn)練的,但 PepNet(和其他 de novo 算法)不受訓(xùn)練樣本的特定肽知識的限制。因此,PepNet 可以用作蛋白質(zhì)組數(shù)據(jù)分析的強(qiáng)大工具,特別是當(dāng)沒有全面的目標(biāo)蛋白質(zhì)序列數(shù)據(jù)庫時(例如,在宏蛋白質(zhì)組學(xué)中)。
研究人員認(rèn)為,高精度肽測序的能力將使從頭肽測序在蛋白質(zhì)組學(xué)數(shù)據(jù)分析中的應(yīng)用不斷增加。除了論文里介紹的 HCD 譜圖肽測序之外,PepNet 還可以擴(kuò)展到使用其他碎片方法獲取的 MS/MS 譜圖,例如電子轉(zhuǎn)移解離(ETD)、電子轉(zhuǎn)移/高能碰撞解離(EThcD)、光解離(PD)和紅外多光子解離(IRMPD)。這些方法通常被認(rèn)為會產(chǎn)生復(fù)雜的 MS/MS 譜圖,其中嵌入復(fù)雜 MS/MS 譜圖的豐富信息有望提高從頭肽測序的準(zhǔn)確性。
總而言之,PepNet 顯著提高了從頭肽測序的準(zhǔn)確性,因此可以作為數(shù)據(jù)庫搜索引擎的補充工具,用于蛋白質(zhì)組學(xué)中的肽鑒定。因此,研究人員認(rèn)為 PepNet 將提高蛋白質(zhì)組學(xué)數(shù)據(jù)分析的效率,并造福生命科學(xué)研究。
Tags:
相關(guān)推薦



- 免責(zé)聲明
- 本文所包含的觀點僅代表作者個人看法,不代表新火種的觀點。在新火種上獲取的所有信息均不應(yīng)被視為投資建議。新火種對本文可能提及或鏈接的任何項目不表示認(rèn)可。 交易和投資涉及高風(fēng)險,讀者在采取與本文內(nèi)容相關(guān)的任何行動之前,請務(wù)必進(jìn)行充分的盡職調(diào)查。最終的決策應(yīng)該基于您自己的獨立判斷。新火種不對因依賴本文觀點而產(chǎn)生的任何金錢損失負(fù)任何責(zé)任。
