

 新火種
2023-10-28
新火種
2023-10-28 


哈工大與騰訊開發:一種專門針對蛋白質組數據設計的反卷積方法

編輯 | 蘿卜皮
細胞類型反卷積是一種用于從大量測序數據中確定/解析細胞類型比例的計算方法,并且經常用于分析腫瘤組織樣本中的不同細胞類型。然而,由于重復性/再現性、參考標準可變以及缺乏單細胞蛋白質組參考數據的挑戰,使用蛋白質組數據分析細胞類型的反卷積技術仍處于起步階段。
哈爾濱工業大學、騰訊 AI lab 以及蘇黎世聯邦理工學院的研究團隊合作開發了一種專門針對蛋白質組數據設計的基于深度學習的反卷積方法(scpDeconv)。
scpDeconv 使用自動編碼器利用來自批量蛋白質組數據的信息來提高單細胞蛋白質組數據的質量,并采用域對抗架構來橋接單細胞和批量數據分布,并將標簽從單細胞數據轉移到批量數據 。
該研究以「Deep domain adversarial neural network for the deconvolution of cell type mixtures in tissue proteome profiling」為題,于 2023 年 10 月 19 日發布在《Nature Machine Intelligence》。

亟需處理單細胞蛋白質組數據的新方法
批量測序技術一般是指對特定組織、器官或器官系統中的所有細胞進行裂解和測序,并隨后測量基因/蛋白質的平均豐度。因此,批量測序忽略了樣品的細胞異質性。單細胞(轉錄組/蛋白質組)測序技術的發展允許從組織中分離單細胞,以分析和測量各個分子。
盡管該技術在概念上具有優勢,但由于成本相對較高且與福爾馬林固定或石蠟包埋組織的樣本不兼容,應用它來探索大樣本隊列中腫瘤微環境的細胞異質性一直具有挑戰性。因此,人們開發了反卷積方法來從大量組織樣本的表達譜中推斷細胞類型及其比例,為臨床背景下研究腫瘤微環境的細胞組成提供了一種相對低成本和便捷的方法。
然而,到目前為止,現有的反卷積方法僅關注轉錄組學。盡管蛋白質組提供了從理解生物機制到發現藥物靶點的關鍵生物學和臨床信息,但尚未關注對大量蛋白質組數據進行解卷積。
此外,由于復雜的轉錄后調控、RNA/蛋白質降解和翻譯后修飾,大多數蛋白質的濃度無法通過相應基因的轉錄/表達水平準確表示。大多數蛋白質的拷貝數比相應轉錄本高 1,000 倍以上,并且表達比轉錄本更廣泛的動態豐度范圍,因此在區分細胞和細胞狀態并提供獨特的生化見解方面表現出更高的潛力。
所以,迫切需要開發基于單細胞蛋白質組數據的新方法來對大量蛋白質組樣本進行解卷積。
已有反卷積方法有諸多挑戰
迄今為止,最常用的單細胞蛋白質組技術主要基于分子標記抗體,但是檢測到的蛋白質數量有限。隨著基于質譜的單細胞蛋白質組學技術的出現,單個細胞中檢測到的蛋白質數量已大幅增加至多達 1,000-3,000 個細胞內蛋白質,從而擴大了檢測到的蛋白質的數量和類型。然而,處理足夠數量的細胞來生成數據以支持來自批量數據的細胞類型反卷積仍然具有挑戰性。
與轉錄組數據相比,訓練和基準反卷積算法所需的單細胞蛋白質組數據還存在許多額外的挑戰。這些包括單細胞蛋白質組數據中的大量背景噪音、較差的數據質量和取決于分析運行和/或技術的顯著變化,以及有限的蛋白質組覆蓋范圍。
此外,現有的針對轉錄組數據設計的反卷積方法并不直接適用于蛋白質組數據。
首先,蛋白質豐度和轉錄本表達有不同的分布和值范圍。其次,與批量蛋白質組學相比,單細胞蛋白質組學檢測到的蛋白質組覆蓋率要低得多,這是現有反卷積方法未考慮的情況。第三,對于單細胞蛋白質組數據和批量蛋白質組數據,「批次效應」(分布之間的變異性)很明顯,這是現有的反卷積方法很少考慮的。
基于深度學習的方法 scpDeconv
為了應對這些挑戰,通過充分考慮最近開發的單細胞蛋白質組技術的固有特征,哈爾濱工業大學、騰訊 AI lab 以及蘇黎世聯邦理工學院的研究團隊開發了一種專門針對蛋白質組數據定制的新的反卷積方法——scpDeconv。
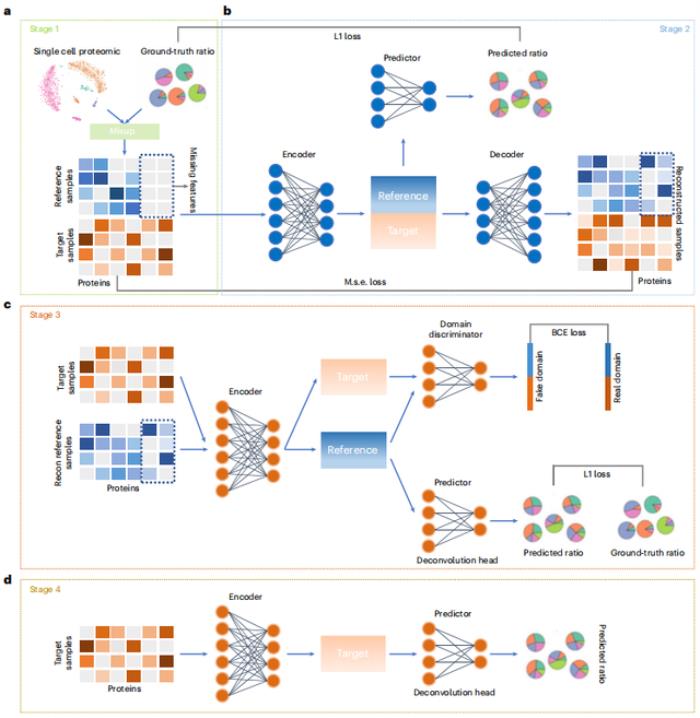
圖示:scpDeconv 方法的架構。(來源:論文)
通過使用自動編碼器網絡,scpDeconv 的插補模塊可以插補單細胞蛋白質組數據中缺失的低豐度蛋白質的值。這是通過對混合的單細胞蛋白質組數據和目標批量蛋白質組數據的聯合學習來實現的。通過領域對抗訓練,scpDeconv 可以提取估算參考數據的領域不變潛在特征并將標簽轉移到目標數據。
該團隊嚴格而廣泛的實驗評估了 scpDeconv 在細胞類型反卷積以及從大量組織蛋白質組數據集中推斷細胞周期狀態方面的性能和魯棒性。此外,對臨床黑色素瘤樣本中現有的大量蛋白質組數據集的重新分析顯示了 scpDeconv 在癌癥診斷和預后中的臨床應用價值。
scpDeconv 的優勢
與現有的針對轉錄組學的反卷積方法相比,scpDeconv 為蛋白質組數據反卷積提供了四個級別的優勢。
(1)scpDeconv 以數據驅動的方式基于參考數據和目標數據的聯合學習來推斷細胞類型比例,因此不受先前數據分布假設的限制,并且與來自不同實驗或技術背景的廣泛蛋白質組數據兼容。
(2)scpDeconv引入了域適應策略,可以有效地處理批次效應并縮小參考數據和目標數據之間的分布差距。
(3)scpDeconv 可以在插補模塊的幫助下插補單細胞蛋白質組數據中低豐度蛋白質的缺失值,并提高參考數據的質量。
(4)研究人員驗證了scpDeconv的輸出能夠反映腫瘤微環境中的細胞類型分布,并且由此確定的細胞類型分布對于疾病診斷和預后有意義。
局限性與展望
然而,scpDeconv 也顯示出一些局限性。模型訓練所需的具有已知細胞類型組成的參考蛋白質組數據,是由具有標記細胞類型的單細胞蛋白質組數據構建的。目前,單細胞蛋白質組學仍處于起步階段,尚未應用于廣泛的生物樣品。該研究中詳盡收集的單細胞蛋白質組數據僅涉及少數細胞類型,從而限制了 scpDeconv 目前的適用性。
該算法專為使用單細胞蛋白質組數據作為參考從蛋白質組數據中進行細胞類型/狀態解卷積而定制,將具有許多關鍵應用。這些包括細胞類型的反卷積、細胞狀態的估計(以細胞周期狀態為例)以及對多種癌癥的微環境的重新分析,從而為已經獲得的大量蛋白質組數據集添加額外的值。
因此,研究人員預計 scpDeconv 將支持從大量蛋白質組數據中提取額外的、生物學和臨床上重要的結果,從而使蛋白質組學界受益。隨著對公開的單細胞蛋白質組圖譜的探索,未來在更廣泛的背景下重新分析現有的組織蛋白質組數據將是可行的。此外,憑借一系列針對多種癌癥類型的可靠微環境參考,相信 scpDeconv 將能夠以低成本分析某些腫瘤樣本的所有成分,從而有利于基礎醫學研究。
論文鏈接:https://www.nature.com/articles/s42256-023-00737-y
相關推薦




- 免責聲明
- 本文所包含的觀點僅代表作者個人看法,不代表新火種的觀點。在新火種上獲取的所有信息均不應被視為投資建議。新火種對本文可能提及或鏈接的任何項目不表示認可。 交易和投資涉及高風險,讀者在采取與本文內容相關的任何行動之前,請務必進行充分的盡職調查。最終的決策應該基于您自己的獨立判斷。新火種不對因依賴本文觀點而產生的任何金錢損失負任何責任。
